




▲第一作者:謝李燕;朱青;張國楨 ;通訊作者:江俊
通訊單位:中國科學技術大學化學與材料科學學院
論文DOI:10.1021/jacs.0c00561
全文速覽
系統闡述了利用金屬與金屬氧化物功函數差異給后者注入電子從而誘導質子遷入氧化物晶格的物理化學機制,并發展了一套成熟的酸-金屬聯合處理應用方案,實現溫和條件下金屬氧化物半導體可控加氫和性質調控。
背景介紹
A加氫是調控材料性能最重要的技術之一
金屬氧化物半導體是一類重要的半導體材料,具有豐富可調的電、光、磁學性質。金屬氧化物氫化處理是調節其性質提升效用的重要方式。例如,氫化TiO2可增強光吸收改進光催化性能(Science 2011, 331, 746),氫化VO2會發生電子相變,拓寬其在光電領域的應用(Nat. Commun. 2018, 9, 818; Sci. Adv. 2019, 5, eaav6815),氫化WO3和MoO3可實現局域表面等離子共振的調控(J. Am. Chem. Soc. 2016, 138, 9316)。
B傳統的加氫方法條件苛刻
傳統的加氫策略用H2作為氫源,但因為H2鍵能高達436 KJ/mol,斷開H-H鍵能耗很大,而且H原子進入金屬氧化物半導體晶格也依然需要高能驅動,因此這個策略需要高溫高壓和貴金屬催化劑,成本很高(Chem. Rev. 2019, 119 , 4777)。與此同時,酸溶液是雖然是方便而廉價的質子源,但利用酸來給金屬氧化物加氫鮮有成功。
研究盲點:酸溶液中的氫質子為什么很少被用在金屬氧化物半導體加氫上
研究出發點
利用酸溶液作為氫源,首先要了解利用酸質子給氧化物加氫的難點在哪。以往的實驗觀察到,金屬氧化物在酸中要么表現為化學惰性(TiO2,WO3, MoO3和Nb2O5)要么會被酸腐蝕(如VO2。而且,直接用酸溶液實現金屬氧化物的加氫需要解決加氫后電荷平衡的問題。這是因為陰離子體積大而無法進入金屬氧化物晶格,僅有氫質子進入金屬氧化物體相將破壞電中性,而且表面靜電相斥作用也阻止氫質子在金屬氧化物體相累積。我們從金屬接觸VO2可以阻止后者被酸腐蝕的實驗現象(Nat. Commun. 2018, 9 , 818)出發,提出了一個大膽的設想,是否可以發展一套用金屬輔助酸溶液中金屬氧化物加氫的策略?于是,我們用第一性原理計算模擬了一批金屬氧化物(TiO2,WO3, MoO3和Nb2O5)的金屬-酸溶液聯合加氫過程,隨后做了實驗對計算結果進行了驗證。
我們的結論是,這個策略不僅可行,而且可以實現可控加氫(即調節氫化的程度)。其關鍵在于兩方面的調控:一、金屬與金屬氧化物功函數差異;二、質子遷移進入金屬氧化物晶格的勢壘。我們隨后對所得加氫產物的光,電,熱,磁等性質變化進行了研究,發現加氫顯著拓寬了金屬氧化物材料性質的可變范圍,為新材料的設計提供了理論指導。
圖文解析
a. 機理圖
這個工作的機理如圖1所示主要分為三步:(1) 功函數的差異驅動電子在金屬/金屬氧化物界面處從金屬流向金屬氧化物,導致金屬氧化物負電荷;(2)金屬氧化物中的負電荷吸引溶液中的質子向晶格擴散;(3) 氫擴散到金屬氧化物晶格后形成了穩定的氫摻雜。
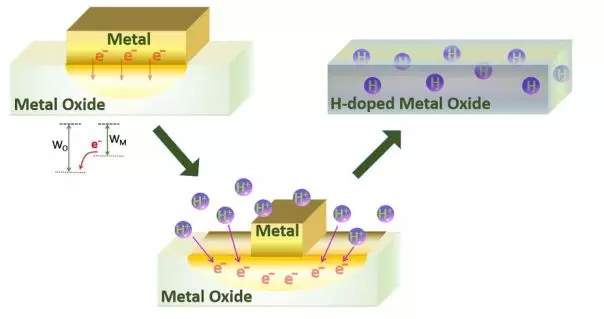
▲圖1:金屬誘導酸中氫質子加入金屬氧化物體相實現金屬氧化物加氫的機理圖。
b. 理論計算部分。
首先,我們以銳鈦礦型TiO2為模型體系來探討這個加氫方法的可行性。我們選擇了金屬Al、Zn、Cu和Ag,它們的功函數(WF)分別是4.11、4.29、4.32和4.44 eV,均低于TiO2(6.67 eV),允許電子從金屬流向TiO2。然后,我們對金屬與TiO2(101)的界面模型來進行差分電荷分析,證實了電子在界面處從金屬轉移到TiO2(圖2)。另外,Bader電荷分析顯示這種界面電荷極化導致平均每個金屬原子有0.02~0.09個負電荷轉移到TiO2,使后者攜帶負電荷,而負電荷與酸溶液中質子的靜電作用將變為氫原子進入晶體的驅動力。
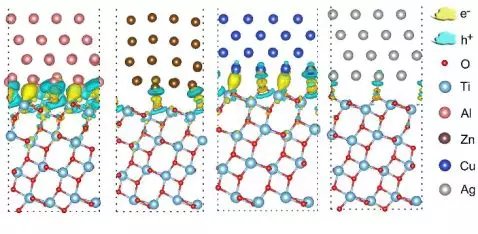
▲圖2:電子從金屬表面轉移到金屬氧化物表面示意圖。黃色和藍色分別代表電荷遷入和遷出的區域。
我們計算位于表面三種不同位置的氫原子遷入TiO2晶格的能壘,如圖3所示。圖3b-d中收集了不同電荷條件(1 h: 1個空穴, 0 e: 中性, 1 e: 1個電子)下質子遷移進其晶格的勢能面。不論質子從哪個位置遷入,其勢壘都隨著體系負電荷的增加而降低,表明銳鈦礦型TiO2中的負電荷累積可促進氫向其晶格的擴散。以從位點1開始遷移(圖3b)為例,在中性條件(0 e)下,氫原子從晶格表面向次表面的遷移必須越過0.97 eV的勢壘并吸收0.22ev的熱量(ΔE)。相比之下,負電荷條件(1e)顯著降低了能壘(0.47ev),并將吸熱反應轉變為放熱反應(ΔE=-0.45ev),而正電荷條件(1h)能壘提高(1.24ev),反應吸熱增加(ΔE=0.45ev)。注:ΔE=末態體系能量-初態體系能量。
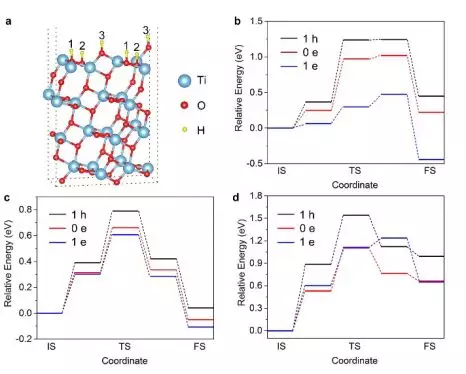
▲圖3:電荷累積在金屬氧化物上導致氫進入TiO2晶格的能壘降低。(a)H在銳鈦礦型TiO2(101)表面的三個可能結合位點,標記為位點1/2/3。從位點1(b)、位點2(c)和位點3(d)開始的氫遷移進入TiO2晶格的能量變化圖。起始位置的能量設置為0 eV。
c. 簡單實驗驗證
作為理論研究為主的課題組,我們嘗試進行簡單實驗,驗證金屬-酸溶液處理方法的氫化效果。用Zn金屬與半導體混合,投入鹽酸溶液中處理,白色的原始銳鈦礦-TiO2迅速轉變為藍黑色(圖4a)。圖4b顯示了1H NMR(核磁共振)光譜數據,指示TiO2晶格中的氫元素信號。如圖4c所示,原始樣品只吸收紫外光,而處理過的樣品在可見光和紅外區域顯示出相當大的光吸收。ESR(電子自旋共振)譜在g=1.94處顯示出強信號(圖4d),這可歸因于傳輸進來的電子大都局域在Ti原子附近,從而產生了Ti3+順磁中心從而被ESR譜觀察到。由于引入了Ti3+中心,摻雜H的TiO2樣品顯示出比原始TiO2更高的室溫鐵磁性(圖4e)。并且理論計算也表明氫化破壞了電子自旋對稱性,并且使費米能級升高實現金屬-絕緣體轉變(metal-insulator transition, MIT)。這將顯著提高電子電荷載流子的濃度。進一步的電學實驗證實,氫化后材料的電導率顯著增加了5個數量級(圖4f)。與此形成鮮明對比的是,高功函數的貴金屬Au/Pd/Pt轉移到TiO2的電荷要少得多。如所料,實驗中的Au/Pt/Pd加酸處理也未能使TiO2氫化。
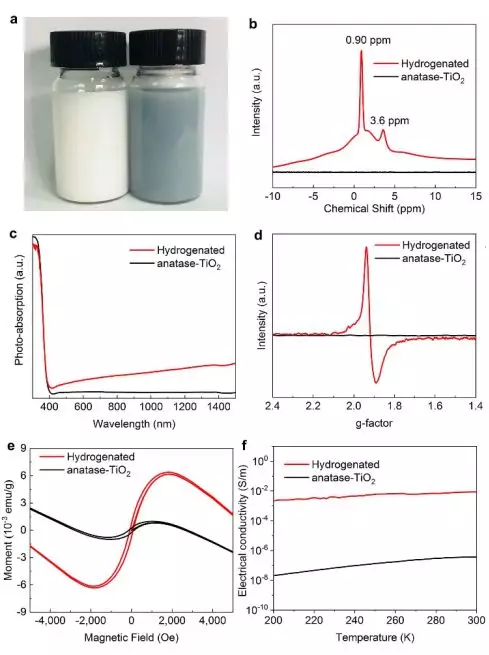
▲圖4:利用Zn-鹽酸處理實現TiO2加氫后材料的性能變化。原始和氫化銳鈦礦型TiO2的(a)照片;(b)1H NMR譜;(c)UV-Vis-NIR吸收光譜;(d)ESR譜;(e)磁滯回線;(f)電導率隨溫度變化圖。
d. 基于理論預測篩選金屬與半導體,有效調控氫化濃度
我們接著用Zn-鹽酸處理實現了Nb2O5,MoO3和WO3的加氫。第一性原理計算表明,這些金屬氧化物具有比Zn更高的功函數,差分電荷分布圖也表明電子會從Zn流入金屬氧化物。如圖5a所示,隨著體系中負電荷的增加,H進入晶格體相的遷移勢壘降低。 圖5b-d 中H信號峰的出現和顏色變深均證實了這些氧化物被有效氫化。圖5e表明加氫氧化物的可見光吸收增強。圖5f所示,加氫導致氧化物電導率顯著增加了4~7個數量級。計算還發現,質子遷移進ZrO2和SnO2體相的能壘非常高,因而不能用電子-質子共摻雜策略實現有效加氫。這一預測也得到了實驗的證實。
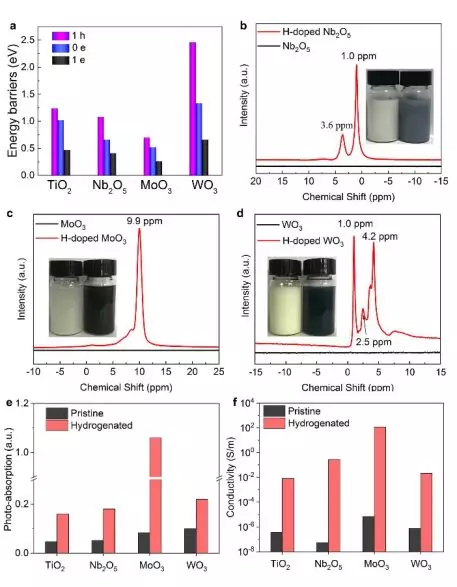
▲圖5:Nb2O5, MoO3和WO3用Zn-鹽酸方法實現加氫的情況。(a) 銳鈦礦型TiO2(1位點)、Nb2O5, MoO3和WO3的H遷移入相應晶格的能壘變化圖。(b)Nb2O5,(c)MoO3和(d)WO3的1H核磁共振譜,插圖是鋅-鹽酸處理前后樣品的照片。Zn-鹽酸處理前后樣品(e)700nm處的光吸收強度和(f)300K處的導電性。
第一性原理計算也發現,和Zn相比,Cu與金屬氧化物的功函數差異更小,因此轉移給氧化物的電荷較少。因此,Cu-鹽酸處理的加氫過程會變慢(圖5a),使金屬氧化物加氫度易于調控。使H擴散勢壘減少較少這使得我們可以很容易地調節H摻雜的程度。如圖6a所示,Cu-鹽酸處理實現了WO3的逐步加氫,控制摻氫濃度。圖6b是我們模擬的不同程度WO3氫化物HxW8O24(x=0~4)的分元素軌道態密度圖。費米能級(0 eV)以下的填充態(陰影區)是電荷載流子,表明氫化后的WO3有準金屬特性。而且,該填充態內的電子數(0.88、1.78、2.72和3.60)隨氫化度增加呈線性增大趨勢,表明氫摻雜引起的載流子濃度變化可用氫摻雜量定量控制。
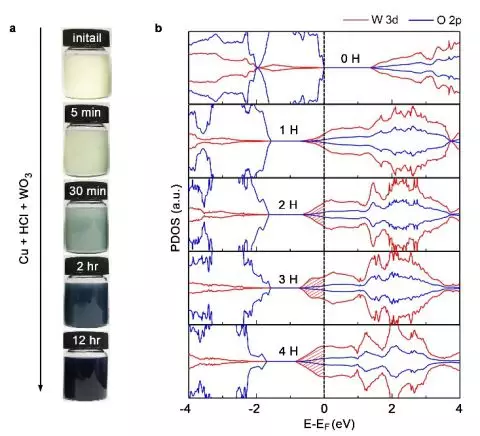
▲圖6:WO3利用Cu-鹽酸處理實現不同程度的加氫。(a)Cu-鹽酸處理WO3后0-12小時的變化。(b)不同H摻雜濃度下WO3的分元素軌道態密度圖(PDOS)。
總結與展望
在這項研究中,通過理論計算,我們確認金屬氧化物氫化過程的熱力學受控于金屬-金屬氧化物功函數差,動力學受控于從金屬轉移到金屬氧化物的負電荷量。通過實驗驗證,我們在溫和條件和酸溶液下實現了若干金屬氧化物(銳鈦礦型TiO2/WO3/MoO3/Nb2O5)的氫摻雜。而且,通過調節功函數差異,我們可以定量控制摻氫濃度,進而調控氫化氧化鎢(HxWO3)的電子結構。我們預計,金屬-酸聯合處理法將成為金屬氧化物可控加氫的一種通用策略,將助力光催化、光學器件和傳感器等領域的新材料的理性設計。
免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。

官方微信
《中國腐蝕與防護網電子期刊》征訂啟事
- 投稿聯系:編輯部
- 電話:010-62316606-806
- 郵箱:fsfhzy666@163.com
- 中國腐蝕與防護網官方QQ群:140808414

“海洋金屬”——鈦合金在艦船的

腐蝕與“海上絲綢之路”