
一文讀懂無機非金屬材料的檢測方法
2022-11-14 15:42:00
作者:測了么測試服務 來源:測了么測試服務
分享至:


無機非金屬材料是以某些元素的氧化物、碳化物、氮化物、鹵素化合物、硼化物以及硅酸鹽、鋁酸鹽、磷酸鹽、硼酸鹽等物質組成的材料。是除有機高分子材料和金屬材料以外的所有材料的統稱。
材料性能不是簡單地由其元素或離子團的成分所決定,而是由這些成分所組成的物相、各物相的相對含量、晶體結構、結構缺陷及分布情況等因素所決定的。為了研究材料的相組成、相結構、相變及結構對性能的影響,確定最佳的配方與生產工藝,必須借助各種檢測設備。本文將以檢測方法為切入點,介紹每種方法在無機非金屬材料的組成與性能研究中的應用。
XRD--物相組成及含量
X射線晶體照射到晶體所產生的衍射具有一定的特征,可用衍射線的方向及強度表征,根據衍射特征來鑒定晶體物相的方法稱為物相分析法。
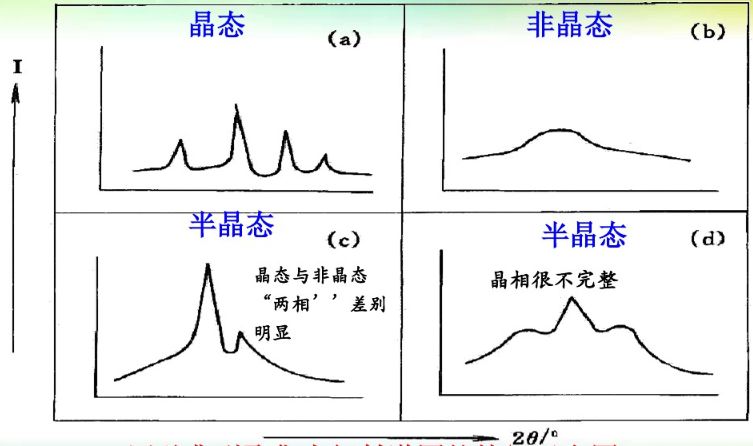
四種典型聚集態衍射譜圖的特征示意圖
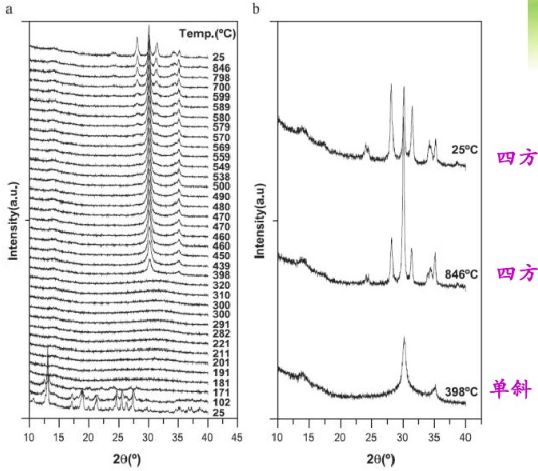
變溫XRD觀察ZrO2的晶型變化過程
XRD樣品制備XRD試樣的制備應考慮晶粒大小、試樣的大小及厚度、擇優取向、加工應變和表面平整度。
粉末樣品的制備一般要求粉末樣品的顆粒度大小在0.1~10μm范圍。將樣品研磨成適合衍射實驗用的粉末,再把樣品粉末制成有一個十分平整平面的試片。
薄膜樣品的制備在薄膜樣品制備時,要求樣品具備比較大的面積,薄膜比較平整以及表面粗糙度要小。對于薄膜樣品,可將其鋸成與窗孔大小一致,然后用橡皮泥直接將其固定在窗孔內,應注意將固定在窗孔內的樣品表面與樣品板齊平。
特殊樣品分散在膠帶紙上黏結,形成石蠟糊,或鋸成與窗孔大小一致,用石蠟固定在窗孔內。
XRD的應用

ICP--物質元素
ICP是電感耦合等離子譜儀。根據檢測器的不同分為ICP-OES(電感耦合等離子發射光譜儀,也稱ICP-AES)和ICP-MS(電感耦合等離子質譜儀)。兩者均能測元素周期表中的絕大部分元素,但能測得元素稍微有異,檢測能力上后者要比前者高。因為ICP光源具有良好的原子化、激發和電離能力,所以它具有很好的檢出限。對于多數元素,其檢出限一般為0.1~100ng/ml,可以同時測試多種元素,靈敏度高,檢測限低,測試范圍寬(低含量成分和高含量成分能夠同時測試)。
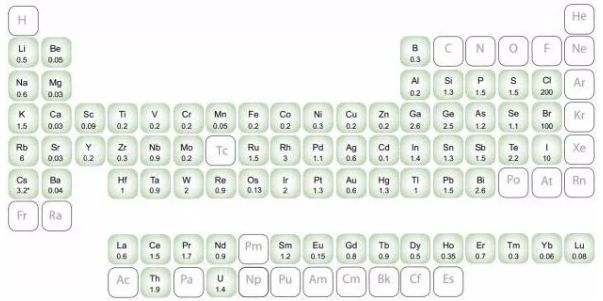
ICP-OES可同時分析常量和痕量組分,無需繁復的雙向觀測,還能同時讀出、無任何譜線缺失的全譜、直讀等離子體發射光譜儀,具有檢出限極低、重現性好,分析元素多等顯著特點,ICP-OES大部份元素的檢出限為1~10ppb,一些元素也可得到亞ppb級的檢出限。ICP-OES的檢測元素如下圖:
ICP-MS電感耦合等離子體質譜儀以質譜儀作為檢測器,通過將樣品轉化為運動的氣態離子并按質荷比(M/Z)大小進行分離并記錄其信息來分析。若其所得結果以圖譜表達,即所謂的質譜圖。ICP-MS的進樣部分及等離子體和ICP-AES的是極其相似的。但ICP-MS測量的是離子質譜,提供在3~250amu范圍內每一個原子質量單位(amu)的信息。還可進行同位素測定。
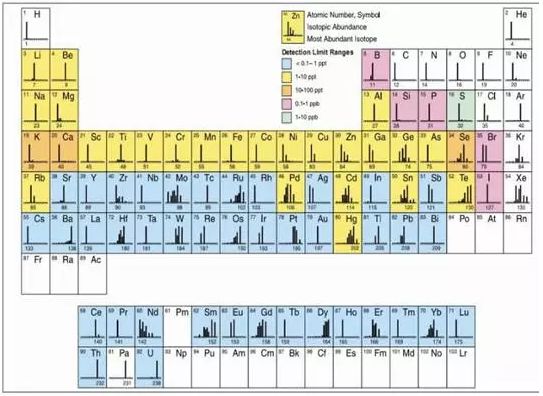
ICP-MS具有極低的檢出限,其溶液的檢出限大部分為ppt級,石墨爐AAS的檢出限為亞ppb級,但由于ICP-MS的耐鹽量較差,ICP-MS的檢出限實際上會變差多達50倍,一些輕元素(如S、Ca、Fe、K、Se)在ICP-MS中有嚴重的干擾,其實際檢出限也很差。ICP-MS的檢測元素和檢測極限如下圖:
XRF--物質元素,定量、定性分析
XRF指的是X射線熒光光譜儀,可以快速同時對多元素進行測定的儀器。在X射線激發下,被測元素原子的內層電子發生能級躍遷而發出次級X射線(X-熒光)。只能測元素單質和氧化物形式。但由于XRF是表面化學分析,故測得的樣品必須滿足很多條件,比如表面光滑、成分均勻。如果成分不均勻,只能說明在XRF測量的那個微區的成分如此,其他的不能表示。
XRF的優點:分析速度高、非破壞性、分析精密度高,固體、粉末、液體樣品等都可以進行分析。測試元素范圍大、可定量分析材料元素組成、分辨率高。
XRF和ICP常用作成分的定量分析,其中XRF用物理方法檢測而ICP用化學方法進行測試。相對XRF,ICP的檢測范圍更寬,檢測極限更低,檢測出的數據更準確。EDS和WDS常用作電鏡的附件進行成分分析,但多作為半定量分析,僅可以看出各個元素的比值和大概分布情況及含量,準確性不如XRF和ICP。
XPS--表面分析
XPS, 全稱為X-ray Photoelectron Spectroscopy(X射線光電子能譜),是一種使用電子譜儀測量X-射線光子輻照時樣品表面所發射出的光電子和俄歇電子能量分布的方法。
XPS可分析什么信息
元素的定性分析:可以根據能譜圖中出現的特征譜線的位置鑒定除H、He 以外的所有元素。
元素的定量分析:根據能譜圖中光電子譜線強度(光電子峰的面積)反應原子的含量或相對濃度。
固體表面分析:包括表面的化學組成或元素組成,原子價態,表面能態分布,測定表面電子的電子云分布和能級結構等。
化合物的結構:可以對內層電子結合能的化學位移精確測量,提供化學鍵和電荷分布方面的信息。
分子生物學中的應用:利用XPS鑒定維生素B12中的少量的Co。
膜表面深度分析:用Ar+離子束對清除材料表面污染層,對材料進行深度剖析。
XPS定性分析規律:氧化作用使內層電子結合能上升,氧化中失電子愈多,上升幅度愈大;還原作用使內層電子結合能下降,還原中得電子愈多,下降幅度愈大;對于給定價殼層結構的原子,所有內層電子結合能的位移幾乎相同。
XPS定性分析鑒定順序:鑒別總是存在的元素譜線,如C、O的譜線;鑒別樣品中主要元素的強譜線和有關的次強譜線;鑒別剩余的弱譜線假設它們是未知元素的最強譜線。
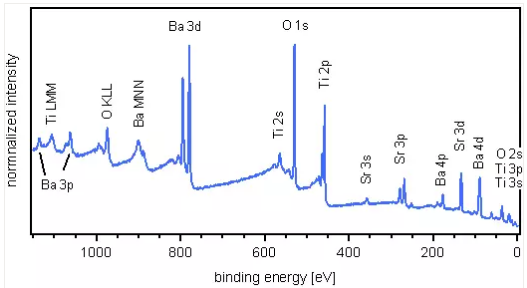
EDS--微區成分分析
EDS是能量色散X射線譜儀,簡稱能譜儀,常用作掃描電鏡或透射電鏡的微區成分分析。利用發射出來的特征X射線能量不同而進行的元素分析,稱為能量色散法。X射線能譜儀的主要構成單元是Si(Li)半導體檢測器,即鋰飄移硅半導體檢測器和多道脈沖分析器。目前還不能用于分析超輕元素(O、N、C等)。由于能譜儀中Si(Li)檢測器的Be窗口吸收超輕元素的X射線,故只能分析Na以后的元素。能譜儀結構簡單,數據穩定性和重現性較好。
EPMA--微區成分分析
電子探針顯微分析儀(EPMA)是掃描成像和成分分析一體化的強大的微區分析工具。
EPMA屬于無損分析,應用廣泛。對任何一種在真空中穩定的固體,均可以用電子探針進行成分分析和形貌觀察,例如金屬、硅酸鹽材料、醫學試樣、纖維、氧化膜、涂層、廢氣顆粒、文物等。尤其在材料科學、地學、礦物學及冶金學等應用最為廣泛。

EPMA的特點及應用:
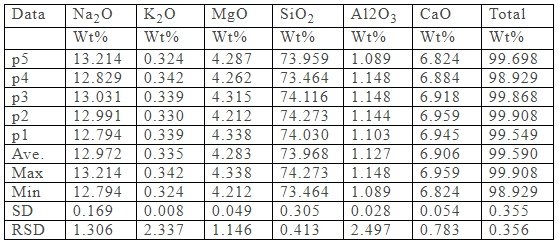
精確定量分析(含超輕元素和微量元素):電子探針是目前微區元素定量分析最準確的儀器。電子探針的檢測極限(能檢測到的元素最低濃度)一般為100-500ppm,不同測量條件和不同物質/元素有不同的檢測極限。在作了基體修正以后,大多數情況下測試結果優于2%。
以硅酸鹽玻璃的定量分析為例,定量分析結果如下,所有的分析結果均在國家標準規定的誤差范圍內。
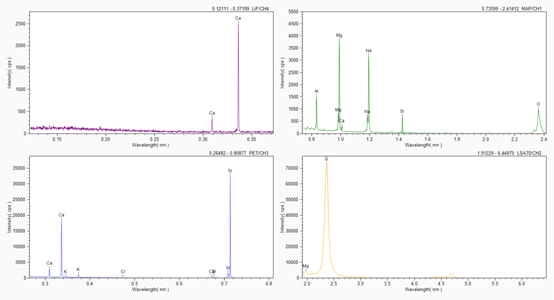
定性分析:主要確定所分析的樣品組成,以硅酸鹽玻璃為例,典型的定性分析結果如下,包括譜圖和歸一化的半定量結果。
線分析:電子束沿一條分析線進行掃描時,能獲得元素及其含量變化的線分布曲線。如果和樣品形貌像對照分析,能直觀地獲得不同元素在不同相或者區域內的分布及變化趨勢。Cu/Cr/Ni/Cu多層結構的線掃描結果如下。
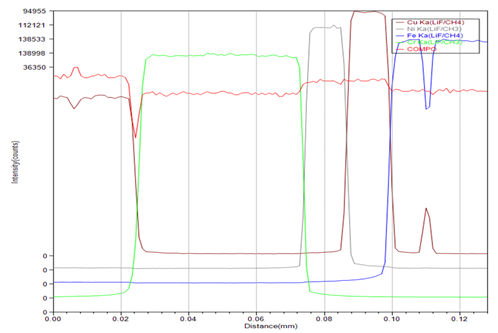
EPMA是微區形貌和成分一體化分析最有效的工具之一,廣泛應用于金屬及合金、半導體、地質、化工、能源、環境、陶瓷、玻璃、稀土氧化物、高分子、生物、醫學、考古、刑偵等各領域。
超高分辨率和超高靈敏度面分析(BES電子像和元素分布圖):EPMA能夠得到納米顆粒、低含量元素和輕元素的面分布圖。對材料晶界、析出相、沉淀物和夾雜相等成分分析有突出優勢,靈敏度和分辨率明顯優于能譜儀。

BSE電子像樣品中的W元素分布圖

BSE電子像樣品中的Sb元素分布圖
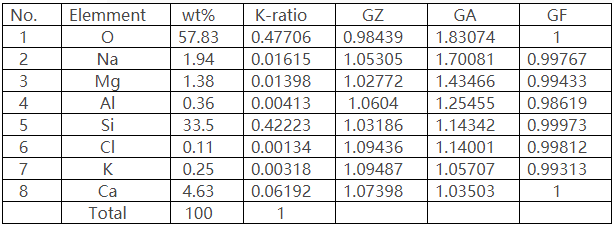
EDS和EPMA上WDS的比較:WDS和EDS對鈦鋁合金中的C元素面分布的測試對比,相同的區域、相同的加速電壓和相同的測試時間:
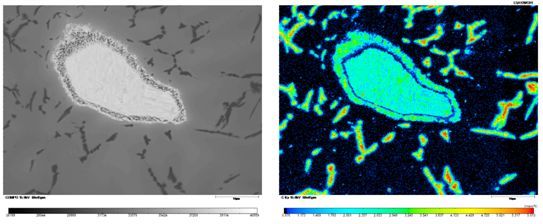
EPMA上WDS測試結果
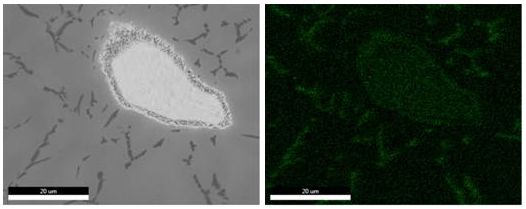
EDS測試結果
紅外光譜--官能團、化學組成
光譜分析是一種根據物質的光譜來鑒別物質及確定它的化學組成,結構或者相對含量的方法。按照分析原理,光譜技術主要分為吸收光譜,發射光譜和散射光譜三種;按照被測位置的形態來分類,光譜技術主要有原子光譜和分子光譜兩種。紅外光譜屬于分子光譜,有紅外發射和紅外吸收光譜兩種,常用的一般為紅外吸收光譜。
紅外吸收光譜是由分子振動和轉動躍遷所引起的, 組成化學鍵或官能團的原子處于不斷振動(或轉動)的狀態,其振動頻率與紅外光的振動頻率相當。所以,用紅外光照射分子時,分子中的化學鍵或官能團可發生振動吸收,不同的化學鍵或官能團吸收頻率不同,在紅外光譜上將處于不同位置,從而可獲得分子中含有何種化學鍵或官能團的信息。
紅外光譜法實質上是一種根據分子內部原子間的相對振動和分子轉動等信息來確定物質分子結構和鑒別化合物的分析方法。只有當振動時,分子的偶極矩發生變化時,該振動才具有紅外活性。
分子的主要振動類型
在中紅外區,分子中的基團主要有兩種振動模式,伸縮振動和彎曲振動。伸縮振動指基團中的原子沿著價鍵方向來回運動(有對稱和反對稱兩種),而彎曲振動指垂直于價鍵方向的運動(搖擺,扭曲,剪式等)。
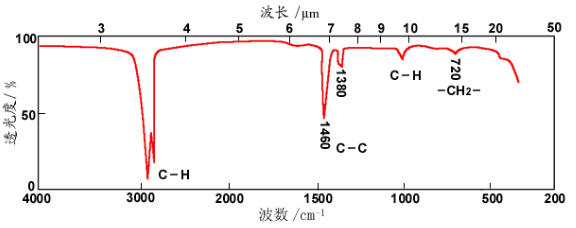
紅外光譜和紅外譜圖的分區
通常將紅外光譜分為三個區域:近紅外區(0.75~2.5 μm)、中紅外區(2.5~25 μm)和遠紅外區(25~300 μm)。一般說來,近紅外光譜是由分子的倍頻、合頻產生的;中紅外光譜屬于分子的基頻振動光譜;遠紅外光譜則屬于分子的轉動光譜和某些基團的振動光譜。(注:由于絕大多數有機物和無機物的基頻吸收帶都出現在中紅外區,因此紅外光譜儀研究和應用最多的區域是中紅外區,積累的資料也最多,儀器技術最為成熟。通常所說的紅外光譜即指中紅外光譜。
按吸收峰的來源,可以將中紅外光譜圖(2.5~25 μm)大體上分為特征頻率區(2.5~7.7 μm,即4000-1330 cm-1)以及指紋區(7.7~16.7μm,即1330-400 cm-1)兩個區域。其中特征頻率區中的吸收峰基本是由基團的伸縮振動產生,數目不是很多,但具有很強的特征性,因此在基團鑒定工作上很有價值,主要用于鑒定官能團。如羰基,不論是在酮、酸、酯或酰胺等類化合物中,其伸縮振動總是在5.9μm左右出現一個強吸收峰,如譜圖中5.9μm左右有一個強吸收峰,則大致可以斷定分子中有羰基。
指紋區的情況不同,該區峰多而復雜,沒有強的特征性,主要是由一些單鍵C-O、C-N和C-X(鹵素原子)等的伸縮振動及C-H、O-H等含氫基團的彎曲振動以及C-C骨架振動產生。當分子結構稍有不同時,該區的吸收就有細微的差異。這種情況就像每個人都有不同的指紋一樣,因而稱為指紋區。指紋區對于區別結構類似的化合物很有幫助。
紅外吸收光譜主要用于定性分析分子中的官能團,也可以用于定量分析(較少使用,特別是多組分時定量分析存在困難)。紅外光譜對樣品的適用性相當廣泛,固態、液態或氣態樣品都能應用,無機、有機、高分子化合物都可檢測。
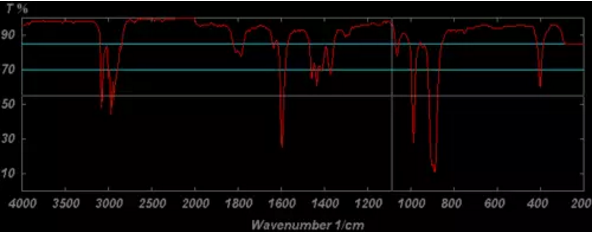
紅外光譜是分子振動光譜,所以萬變不離其宗,紅外光譜測試無機物和有機物是一樣的,都是研究在振動中伴隨有偶極矩變化的基團。常見的所研究的無機物主要包括H2O, CO, 氧化物,無機鹽中的陰離子,配位化合物等。對于無機鹽而言,陽離子類型不同會影響到其陰離子的振動頻率。例如,對于無水堿性氫氧化物而言,OH-的伸縮振動頻率都在3550-3720 cm-1范圍內。其中,KOH為3678 cm-1,NaOH在3637 cm-1, Mg(OH)2為3698 cm-1,Ca(OH)2為3644 cm-1。
常見無機物中陰離子在紅外區的吸收頻率如下表所示:

拉曼光譜
光照射到物質上發生彈性散射和非彈性散射。彈性散射的散射光是與激發光波長相同的成分。非彈性散射的散射光有比激發光波長長的和短的成分, 統稱為拉曼效應。
拉曼散射光譜的特征:拉曼散射譜線的波數雖然隨入射光的波數而不同,但對同一樣品,同一拉曼譜線的位移與入射光的波長無關,只和樣品的振動轉動能級有關。在以波數為變量的拉曼光譜圖上,斯托克斯線和反斯托克斯線對稱地分布在瑞利散射線兩側, 這是由于在上述兩種情況下分別相應于得到或失去了一個振動量子的能量。一般情況下,斯托克斯線比反斯托克斯線的強度大。這是由于Boltzmann分布,處于振動基態上的粒子數遠大于處于振動激發態上的粒子數。
拉曼光譜的應用方向:
拉曼光譜分析技術是以拉曼效應為基礎建立起來的分子結構表征技術,其信號來源與分子的振動和轉動。拉曼光譜的分析方向有:
定性分析:不同的物質具有不同的特征光譜,因此可以通過光譜進行定性分析。
結構分析:對光譜譜帶的分析,又是進行物質結構分析的基礎。
定量分析:根據物質對光譜的吸光度的特點,可以對物質的量有很好的分析能力。
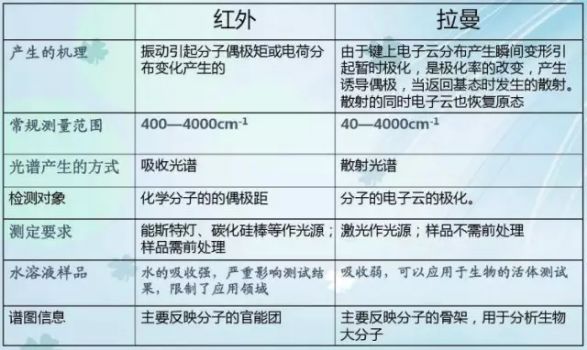
紅外光譜與拉曼光譜的異同點
相同點:對于一個給定的化學鍵,其紅外吸收頻率與拉曼位移相等,均代表第一振動能級的能量。因此,對某一給定的化合物,某些峰的紅外吸收波數和拉曼位移完全相同,紅外吸收波數與拉曼位移均在紅外光區,兩者都反映分子的結構信息。拉曼光譜和紅外光譜一樣,也是用來檢測物質分子的振動和轉動能級。
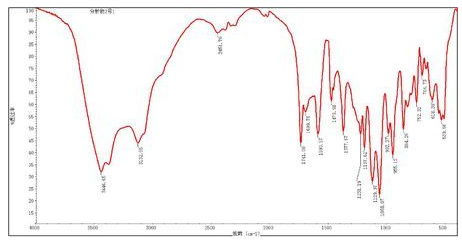
紅外光譜圖
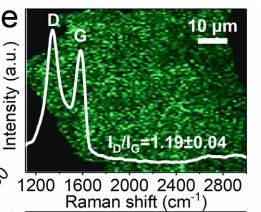
石墨烯基材料的拉曼
區別:
紫外光譜--鑒別、雜質檢查和定量測定
光照射樣品分子或原子時,外層電子吸收一定波長紫外光,由基態躍遷至激發態而產生的光譜。
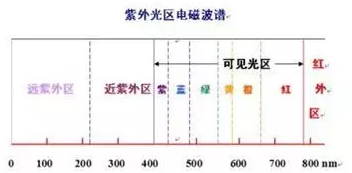
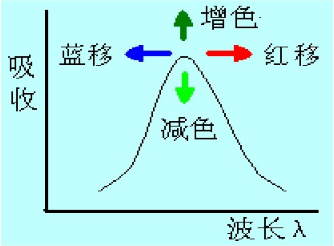
紫外光譜中的特征吸收峰:1、生色基:分子中能夠產生電子吸收的不飽和共價基團,成為生色基,產生n->π*、π→π*躍遷電子體系,如:C=C、C=O、-NO2 等;2、助色基:本身是飽和基團(常含雜原子),當連接一個生色團后,則使生色團的吸收波長變長或吸收強度增加(或同時兩者兼有),一般為帶有孤對電子的原子或原子團 。如-OH , -NH2,-Cl等。3、增色與減色效應、紅移與藍移、溶劑效應:使吸收帶峰的吸收強度增加的效應,為增色效應;反之,使吸收帶峰的吸收強度減弱的效應,為減色效應。由于取代基或溶劑的影響使吸收峰向長波或短波方向移動,最大吸收峰向長波長方向移動為紅移,最大吸收峰向短波長方向移動為藍移。溶劑極性不同會引起化合物吸收峰產生紅移或藍移,稱為溶劑效應。
紫外光譜的分析應用:主要應用于共軛體系(共軛烯烴、不飽和羰基化合物)以及芳香族化合物的分析。吸收譜帶少,吸收譜帶寬。一般來說,利用紫外吸收光譜進行定性分析信號較少。
熒光光譜--光電檢測
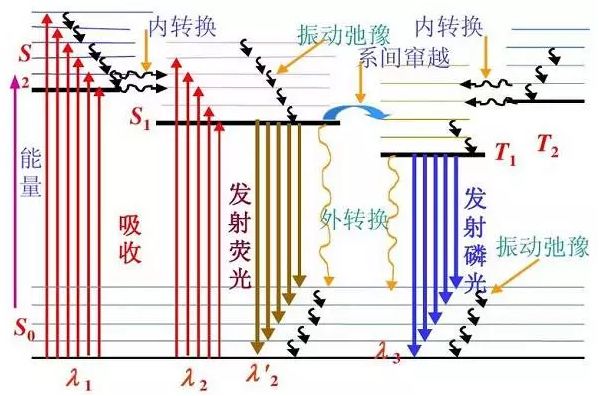
分子的激發與失活過程
任何熒光化合物都具有兩個特征光譜:激發光譜和發射光譜。激發光譜反映了某一固定的發射波長下所測量的熒光強度對激發波長的依賴關系;發射光譜反映了某一固定激發波長下所測量的熒光的波長分布。
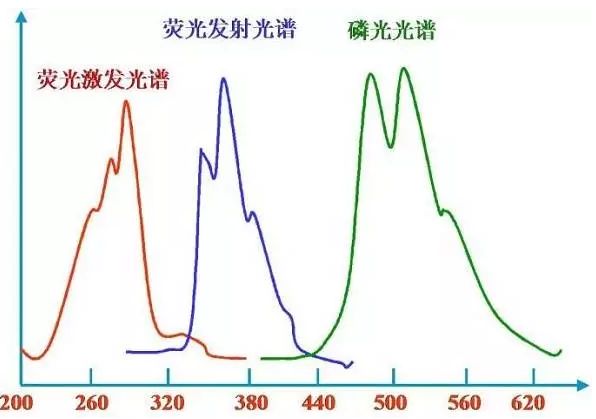
室溫下菲的乙醇溶液的激發光譜和熒(磷)光光譜
熒光光譜能夠提供激發譜、發射譜、峰位、峰強度、量子產率、熒光壽命、熒光偏振度等信息,熒光分析定性和定量的基礎。
熒光光譜的特點:Stokes位移,激發光譜與發射光譜之間有波長差,發射光譜波長比激發光譜波長長。發射光譜的形狀與激發波長無關。鏡像規則,熒光發射光譜與它的吸收光譜成鏡像對稱關系。
熒光物質具有兩個重要的發光參數:熒光壽命和熒光量子產率。熒光壽命(τ)是指當激發停止后,分子的熒光強度降到激發時最大強度的1/e所需的時間,它表示粒子在激發態存在的平均時間,通常稱為激發態的熒光壽命。與穩態熒光提供一個平均信號不同,熒光壽命提供的是激發態分子的信息,前者可以告訴你事情發生了,而后者可以告訴你為什么發生。
熒光分析就是基于物質的光致發光現象而產生的熒光的特性及其強度進行物質的定性和定量的分析方法。目前,也廣泛地作為一種表征技術來研究體系的物理、化學性質及其變化情況,例如生物大分子構象及性質的研究。
熒光光譜適用于固體粉末、晶體、薄膜、液體等樣品的分析。根據樣品分別選配石英池(液體樣品)或固體樣品架(粉末或片狀樣品)。
熒光分析的優點:
(1)靈敏度高;
(2)選擇性強;
(3)試樣量少、方法簡單;
(4)提供較多的物理參數。但是也存在應用范圍不夠廣泛、對環境敏感(干擾因素多)等缺點。
熒光光譜定性分析依據?
不同結構熒光化合物都有特征的激發光譜和發射光譜,因此可以將熒光物質的激發光譜與發射光譜的形狀、峰位與標準溶液的光譜圖進行比較,從而達到定性分析的目的。
熒光光譜定量分析原理?
在低濃度時,溶液的熒光強度與熒光物質的濃度成正比:F=Kc。其中,F為熒光強度,c為熒光物質濃度,K為比例系數。這就是熒光光譜定量分析的依據。
應用領域:熒光光譜分析可與顯微鏡耦合,獲得微區分析結果。熒光是無損傷、非接觸的分析技術,還可用于自動檢驗、批量篩分、遠程原位分析和活體分析。熒光光譜已應用于很多不同領域,特別是需要無損、顯微、化學分析、成像分析的場合。無論是需要定性還是定量的數據,熒光分析都能快速、簡便地提供重要信息。
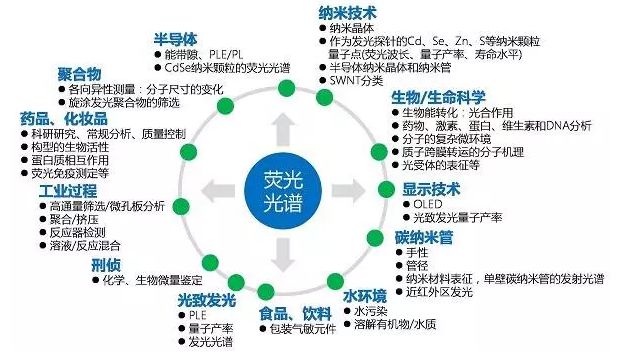
熒光光譜的應用領域
工業分析--水分、灰分、揮發分、固定碳
GB/T 212-2008由中華人民共和國國家質量監督檢驗檢疫總局和中國國家標準化管理委員會發布,是煤的工業分析方法國家標準。該標準規定了煤和水煤漿的水分、灰分和揮發分的測定方法和固定碳的計算方法。在此進行簡單介紹。
水分測試:稱取一定量的一般分析試驗煤樣,置于(105-110)℃干燥箱中,在干燥氣流中干燥到質量恒定。根據煤樣的質量損失計算出水分的質量分數。根據氣流的不同(氮氣或空氣)水分測試可以分為通氮干燥法和空氣干燥法。其中,通氮干燥法應用于所有煤種;空氣干燥法僅適用于煙煤和無煙煤。

灰分測試--無機物含量
灰分是指食品經高溫灼燒后殘留下來的無機物又稱礦物質(氧化物或無機鹽類)。
樣品在灰化時發生了一系列的變化:(1)水分、揮發元素如Cl、I、Pb等揮發散失P、S等以含氧酸的形式揮發散失使無機成分減少。(2)某些金屬氧化物會吸收有機物分解產生的二氧化碳形成碳酸鹽,又使無機成分增多。因此,將灼燒后的殘留物稱為粗灰分。
總灰分的測定原理:把一定量的樣品經碳化后放入高溫爐內灼燒,使有機物被氧化分解,以二氧化碳、氮的氧化物及水等形式逸出,而無機物質以硫酸鹽、磷酸鹽、碳酸鹽、氯化物等無機鹽和金屬氧化物的形式殘留下來,這些殘留物即為灰分,稱量殘留物的重量即可計算出樣品中總灰分的含量。
國標中的方法:稱取一定量的一般分析試驗煤樣品,放入馬弗爐中,以一定的速度加熱到(815±10℃),灰化并灼燒到質量恒定。以殘留物的質量占煤質量的質量分數作為煤樣的灰分。
揮發分:
揮發分(V):煤在高溫和隔絕空氣的條件下加熱時,所排出的氣體液體狀態的產物稱為揮發分。焦炭揮發分是評價焦炭成熟的標志。揮發分的主要成分是甲烷、氫及其他碳氫化合物等。它是鑒別煤炭類別和質量的重要指標之一。一般來講,隨著煤炭變質程度的增加,煤炭揮發分降低。褐煤、氣煤揮發分較高,瘦煤、無煙煤揮發分較低。
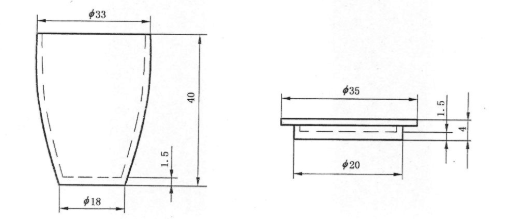
揮發分坩堝

水分測試
卡爾費休法簡稱費休法,是1935年卡爾費休(KarlFischer)提出的測定水分的容量分拆方法。費休法是測定物質水分的各類化學方法中,對水最為專一、最為準確的方法。雖屬經典方法但經過近年改進,提高了準確度,擴大了測量范圍,已被列為許多物質中水分測定的標準方法,主要用于測試物料含水率。
卡爾費休法原理:
費休法屬碘量法,其基本原理是利用碘氧化二氧化硫時,需要-定量的水參加反應。
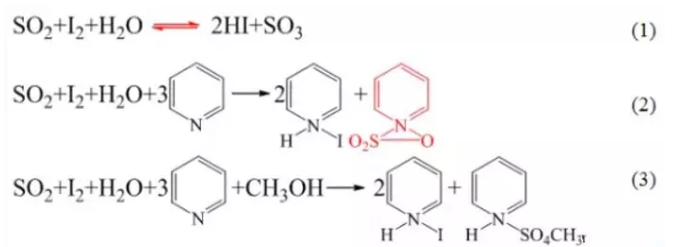
卡爾費休法基本原理為式(1),即碘將二氧化硫氧化為三氧化硫過程需要水分的參與。
注意,式(1)反應為可逆反應,為使其向正反應方向進行完全,在卡氏液中加入無水吡啶,使得式(1)反應產物HI與SO3被定量吸收,形成氫碘酸吡啶和硫酸酐吡啶,如式(2)。
然而,式(2)反應產物硫酸酐吡啶不穩定,可與水發生副反應,因此加入無水甲醇,形成穩定的甲基硫酸氫吡啶,如式(3)。吡啶和甲醇還可作為反應溶劑。
卡式滴定分類:

卡爾滴定的終點指示方法
溶液顏色變化:
(1)淺黃色變為紅棕色;
(2)電化學方法
終點前,卡式試劑中的I2與水反應消耗掉;;到達終點后,碘過量。指示電極間發生下述電極反應而產生電流:I2=2I--2e(可逆電對)

卡爾費休法優缺點:
優點:分析快速,單次測定通常在5min以內;適用范圍廣;專屬性良好;精度高。
缺點:對環境要求較高,一般要求環境溫度為25℃,濕度不超過60%;卡式液含有吡啶等有毒試劑。
卡式水分儀測定法適用范圍:液體、氣體、固體
適用于各種形態的樣品,可加入適當地萃取溶劑加速水分的釋放。
一般要求加入的樣品消耗卡氏滴定儀滴定管體積的10%~90%的滴定液,測量結果較為準確。
卡氏滴定過程最佳pH范圍為5~7,若因強酸強堿性化合物的加入使得滴定液pH發生顯著改變,可通過加入緩沖鹽改善。若樣品與卡氏液中的碘等化合物發生副反應,可通過干燥爐加熱間接將水分帶入滴定液中,避免樣品直接加入到滴定液。
樣品要求:
液體最好10 ml;固體最好0.2 g以上,建議干燥成粉末測試。
SEM--形貌觀察
掃描電鏡(SEM)是利用聚焦的高能電子束對樣品進行掃描檢測,二次電子激發出高能電子信號,通過對這些高能電子信號的接受、放大和顯示成像,最終獲得試樣表面清晰的組織形貌。
掃描電鏡主要應用:能夠觀察納米結構材料。所謂納米結構材料就是指組成材料的顆粒或結構在0.1~100nm范圍內,在保持表面潔凈的條件下加壓成型而得到的固體材料。
能夠直接觀察大試樣的表面形態。試樣的觀察尺寸可以達到直徑100mm×高50mm,或更大尺寸的試樣,而且對試樣的形狀沒有任何限制。
在觀察厚試樣時,能得到高的分辨率和真實的表面形貌。能夠進行動態觀察。在掃描電鏡中成像的信息主要是電子信息,根據近代電子工業技術水平,即使高速變化的電子信息,也能毫不困難的及時接收、處理和儲存,故可進行一些動態過程的觀察。從試樣表面形貌能夠獲得多方面信息。
掃描電鏡除了觀察表面形貌外,還能進行成分和元素的分析,以及通過電子通道花樣進行結晶學分析,選區尺寸可以從10μm到3μm。
TEM形貌觀察
在最近的十幾年中,透射電子顯微鏡(TEM)對材料學科的發展起到了巨大的推動作用。許多新型的納米材料、材料結構和性能之間的關聯、材料物理化學反應機理等研究成果不斷涌現。這一方面歸功于透射電鏡分辨率(能量分辨率、空間分辨率等)的不斷提升,另一方面則受益于原位電鏡、冷凍電鏡、球差校正電鏡等新技術的相繼出現。
催化材料:一般TEM樣品只能在真空環境下進行表征,無法直接觀察多相催化體系中催化材料的動態變化過程。通過原位氣氛TEM技術實現的環境透射電子顯微鏡(ETEM)則可以直接觀察暴露在液體或者氣體環境下的材料結構,這為開發高性能的多相催化材料、提高催化材料的使用壽命、研究催化反應機理提供了巨大便利。

圖1. 利用TEM觀察Rh/TiO2催化劑
新材料結構表征:TEM最廣泛的用途是對材料結構的表征,但是二維圖像不能直觀反映材料三維空間構造。三維透射電鏡(3D-TEM)是將透射電子衍射與計算機圖像處理相結合而形成的一種材料三維重構方法。應用3D-TEM可以實現對復雜自組裝納米結構的準確表征。
美國康奈爾大學Ulrich Wiesner研究團隊結合冷凍電鏡和3D-TEM,實現了高度對稱、超小尺寸、十二面體無機納米籠的普適性自組裝。結果如圖6所示,該工作確認了十二面體無機納米籠結構的存在。這項研究為硅基無機納米材料的構筑提供了全新的思路。
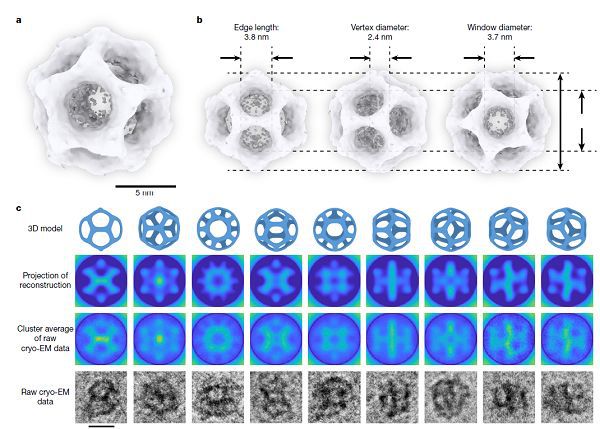
圖6. 十二面體SiO2納米籠的TEM表征與3D重構
當然,TEM所應用的領域遠不只以上幾種:TEM與能譜儀結合(電子能量損失譜等)可以表征出晶格元素價態;原位拉伸TEM可實現對材料力學性能與結構關系的表征;通過TEM可實現原位納米器件的加工,TEM已成為材料學研究中不可或缺的重要手段。
透射電鏡的樣品制備對之后樣品的分析觀察起著至關重要的作用。透射電鏡是利用樣品對入射電子的散射能力的差異而形成襯度,這要求制備出對電子束“透明”的樣品,并要求保持高的分辨率和不失真。對于100~200kV的透射電鏡,要求樣品的厚度為50~100nm,做透射電鏡高分辨率,樣品厚度要求約15nm。
這種方法比上述無機材料樣品的超薄切片法復雜,但是可用于觀察生物的精細結構。需要經過取材、固定、漂洗、脫水、浸透、包埋、切片及染色等多個步驟。
(球差)TEM--原子像
不完美透鏡導致的直接結果就是引入了讓顯微學者最頭疼的球差。電子的聚集是靠洛倫茲力來完結的,在洛倫茲力的效果下,電子以旋進的方法聚集。在TEM里有一條光軸,就和光學顯微鏡中的光軸一樣,違背光軸時,透鏡對光的聚集才能和靠近光軸的聚集才能是不同的。
簡單的說就是磁透鏡在聚集違背光軸的電子時聚集的太厲害了,導致違背光軸的電子束提前在光軸上完結聚集,也就是說實踐聚集點在光軸上面連成了一條線,當這些聚集的電子束在散開去像平面成像的時分,原本應該是一個很小的斑,此時變成了一個很大的斑。無論是在TEM還是在SEM中,尋求更小的聚光斑是永久的方針。尤其是在STEM中,試想,要構成原子像,你總不會期望用一個比原子大的斑去照原子吧?兩個原子之間的間隔大概0.25nm,你一個斑就0.8nm,那么在不考慮衍射的情況下,相鄰的兩個斑都會重合了,更不要說加上衍射了。

球差電鏡還配有EDX和EELS
EDX(Energy-dispersive X-ray spectroscopy)可以用于元素分析,尤其善于重元素的分析。與EDX相比,EELS對于輕元素分辨效果更好,能量分辨率也好出1-2個數量級。由于電子能量損失譜電子伏甚至亞電子伏的分辨率,它可以用于元素價態分析。電子能量損失譜(EELS)也許最適合從碳到3d過渡金屬(從鈧到鋅)的元素分析。
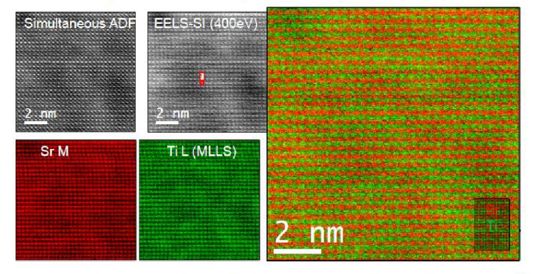
STEM與EELS的結合進行原子級別的結構分析
AFM
原子力顯微鏡(Atomic Force Microscope,AFM):是一種可以用來研究包括絕緣體在內的固體材料表面結構的分析儀器。它將一個對微弱力極敏感的微懸臂一端固定,另一端有一微小針尖,使之與樣品表面輕輕接觸。通過檢測出偏轉量并作用反饋控制其排斥力的恒定,就可以獲得微懸臂對應于各點的位置變化,從而獲得樣品表面形貌的圖像。
原子力顯微鏡在無機非金屬材料的應用:
三維形貌觀測
通過檢測探針與樣品間的作用力可表征樣品表面的三維形貌,這是AFM最基本的功能。AFM 在水平方向具有0.1-0.2nm 的高分辨率,在垂直方向的分辨率約為0.01nm。盡管AFM 和掃描電子顯微鏡(SEM)的橫向分辨率是相似的,但AFM 和SEM 兩種技術的最基本的區別在于處理試樣深度變化時有不同的表征。由于表面的高低起伏狀態能夠準確地以數值的形式獲取,AFM 對表面整體圖像進行分析可得到樣品表面的粗糙度、顆粒度、平均梯度、孔結構和孔徑分布等參數,也可對樣品的形貌進行豐富的三維模擬顯示,使圖像更適合于人的直觀視覺。下圖就是接觸式下得到的二氧化硅增透薄膜原子力圖像,同時還可以逼真的看到其表面的三維形貌。
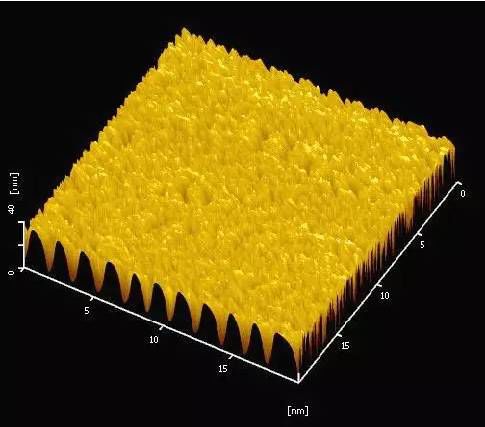
二氧化硅增透薄膜AFM圖
在半導體加工過程中通常需要測量高縱比結構,像溝槽和孔洞,以確定刻蝕的深度和寬度。這些在SEM 下只有將樣品沿截面切開才能測量。AFM 可以無損的進行測量后即返回生產線。下圖為光柵的AFM 圖像,掃描范圍為4×4μm。根據圖的結果,通過profile 功能就可以定量測量刻槽的深度及寬度。
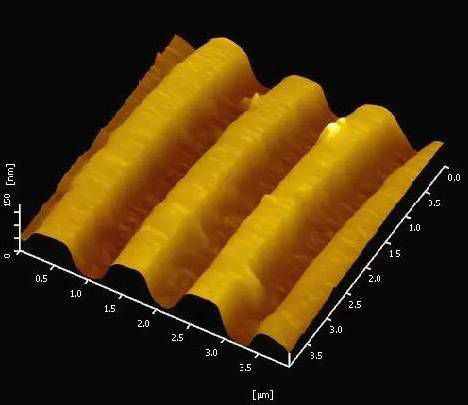
光柵的AFM 圖
AFM 是利用樣品表面與探針之間力的相互作用這一物理現象,因此不受STM 等要求樣品表面能夠導電的限制,可對導體進行探測,對于不具有導電性的組織、生物材料和有機材料等絕緣體,AFM 同樣可得到高分辨率的表面形貌圖像,從而使它更具有適應性,更具有廣闊的應用空間。AFM 可以在真空、超高真空、氣體、溶液、電化學環境、常溫和低溫等環境下工作,可供研究時選擇適當的環境,其基底可以是云母、硅、高取向熱解石墨、玻璃等。AFM 已被廣泛地應用于表面分析的各個領域,通過對表面形貌的分析、歸納、總結,以獲得更深層次的信息。
TG
熱重分析法(Thermogravimetry Analysis,簡稱TG或TGA)為使樣品處于一定的溫度程序(升/降/恒溫)控制下,觀察樣品的質量隨溫度或時間的變化過程,獲取失重比例、失重溫度(起始點,峰值,終止點…)、以及分解殘留量等相關信息。
TG方法廣泛應用于塑料、橡膠、涂料、藥品、催化劑、無機材料、金屬材料與復合材料等各領域的研究開發、工藝優化與質量監控。可以測定材料在不同氣氛下的熱穩定性與氧化穩定性,可對分解、吸附、解吸附、氧化、還原等物化過程進行分析,包括利用TG測試結果進一步作表觀反應動力學研究。可對物質進行成分的定量計算,測定水分、揮發成分及各種添加劑與填充劑的含量。
熱分析的基本原理:
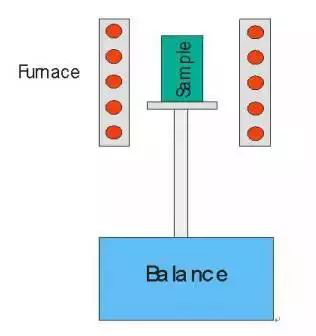
頂部裝樣式的熱重分析儀結構示意圖
爐體為加熱體,在一定的溫度程序下運作,爐內可通以不同的動態氣氛(如N2、Ar、He等保護性氣氛,O2、air等氧化性氣氛及其他特殊氣氛等),或在真空或靜態氣氛下進行測試。
在測試進程中樣品支架下部連接的高精度天平隨時感知到樣品當前的重量,并將數據傳送到計算機,由計算機畫出樣品重量對溫度/時間的曲線(TG曲線)。當樣品發生重量變化(其原因包括分解、氧化、還原、吸附與解吸附等)時,會在TG曲線上體現為失重(或增重)臺階,由此可以得知該失/增重過程所發生的溫度區域,并定量計算失/增重比例。若對TG曲線進行一次微分計算,得到熱重微分曲線(DTG曲線),可以進一步得到重量變化速率等更多信息。典型的熱重曲線如下圖所示:
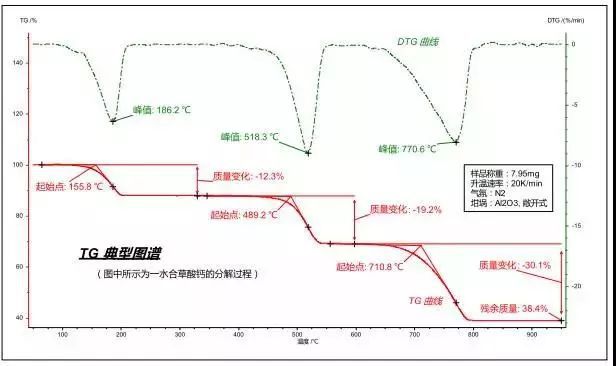
DTG曲線峰值:質量變化速率最大的溫度/時間點,對應于TG曲線上的拐點。
質量變化:分析TG曲線上任意兩點間的質量差,用來表示一個失重(或增重)步驟所導致的樣品的質量變化。
殘余質量:測量結束時樣品所殘余的質量。
另外,在軟件中還可對TG曲線的拐點(與DTG峰溫等同)、DTG曲線外推起始點(更接近于真正意義上的反應起始溫度)、DTG曲線外推終止點(更接近于真正意義上的反應結束溫度)等特征參數進行標示。
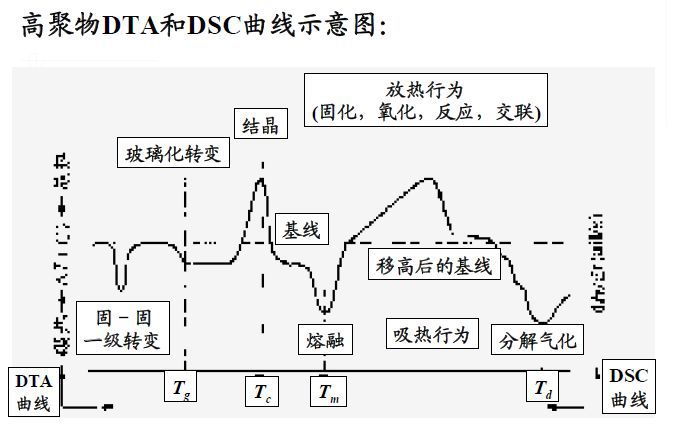
DSC
DSC基本原理與經典應用:在程序溫度(升/降/恒溫及其組合)過程中,測量樣品與參考物之間的熱流差,以表征所有與熱效應有關的物理變化和化學變化。
典型應用:
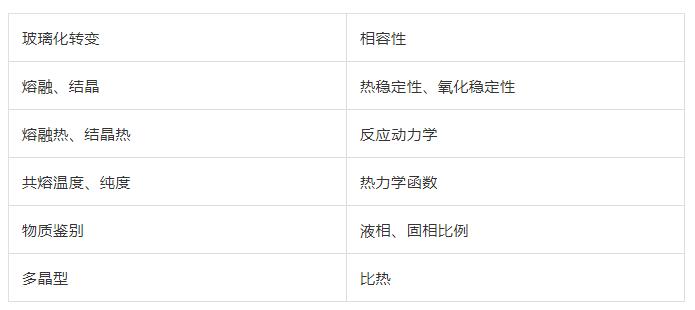
差熱分析(DTA)
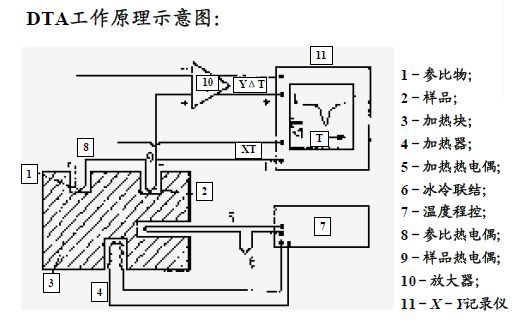
原理:在程序控溫條件下,測量試樣與參比基準物質之間的溫度差與環境溫度的函數關系。
操作方法:用兩個尺寸完全相同的白金坩堝,一個裝參比物,另一個裝待測樣品;將兩只坩堝放在同一條件下受熱,熱量通過試樣容器傳導到試樣內,使其溫度升高,在試樣內會形成溫度梯度;溫度的變化方式會依溫度差熱電偶接點處的位置而不同;溫點插入試樣和參比物中,或坩堝外的底部。
差熱分析曲線
DTA曲線:以溫度為橫坐標、試樣和參比物的溫差ΔT為縱坐標,以不同的吸熱和放熱峰顯示樣品受熱時的不同熱轉變狀態。
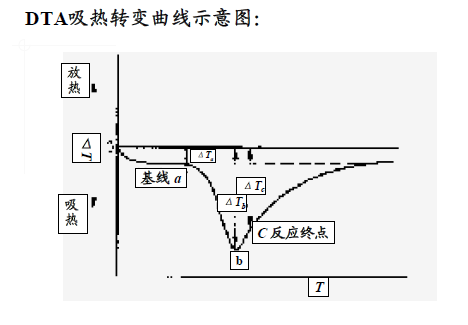
DTA的應用
凡是在加熱(或冷卻)過程中,因物理-化學變化而產生吸熱或者放熱效應的物質,均可以用差熱分析法進行分析。其主要應用范圍如下:
水:對于含吸附水、結晶水或者結構水的物質,在加熱過程中失水時,發生吸熱作用,在差熱曲線上形成吸熱峰。
氣體:碳酸鹽、硫酸鹽及硫化物等,在加熱過程中由于CO2、SO2等氣體的放出而形成吸熱峰。不同類物質放出氣體的溫度不同,差熱曲線的形態也不同,利用這種特征就可以對不同類物質進行區分鑒定。
價態變化材料中含有變價元素,在高溫下由低價態變為高價態而放出熱量,形成放熱峰。變價元素不同,以及在晶格結構中的情況不同,則因氧化而產生放熱效應的溫度也不同。如Fe2+在340~450 ℃變成Fe3+
重結晶某些非晶態物質在加熱過程中會發生重結晶而放出熱量,在差熱曲線上形成放熱峰;晶態物質在加熱過程中晶格結構被破壞,變為非晶態物質發生晶格重構,則也形成放熱峰。
晶型轉變有些物質在加熱過程中由于晶型轉變而吸收熱量,在差熱曲線上形成吸熱峰。
DSC和DTA的區別:DSC的前身是差熱分析DTA,DTA只能測試△T信號,無法建立△H與△T之間的聯系;而DSC測試△T信號,并建立△H與△T之間的聯系。兩種方法的物理含義不一樣,DTA僅可以測試相變溫度等溫度特征點,DSC不僅可以測相變溫度點,而且可以測相變時的熱量變化。DTA曲線上的放熱峰和吸熱峰無確定物理含義,而DSC曲線上的放熱峰和吸熱峰分別代表放出熱量和吸收熱量。DTA曲線上凸表示樣品的溫度比參比樣品的溫度高,下凹表示樣品的溫度比參比樣品的溫度低。DSC曲線上凸表示有熱量釋放出來,下凹表示有熱量吸收,兩者的趨勢應該是大致一樣。
熱導分析
導熱系數是表示材料熱傳導能力大小的物理量。導熱系數也叫導熱率,是指在穩定傳熱條件下,1m厚的材料,兩側表面的溫差為1度(K,℃),在1秒鐘內(1S),通過1平方米面積傳遞的熱量,單位為瓦/米·度 (W/(m·K),此處為K可用℃代替)。
熱擴散系數是表示材質均溫能力大小的物理量。熱擴散系數又叫導溫系數,它表示物體在加熱或冷卻中,溫度趨于均勻一致的能力,單位為平方米/秒( m2/s) 。在導熱系數高的物質中熱能擴散的很快,而導熱系數低的物質中熱能則擴散的較慢。這個綜合物性參數對穩態導熱沒有影響,但是在非穩態導熱過程中,它是一個非常重要的參數。
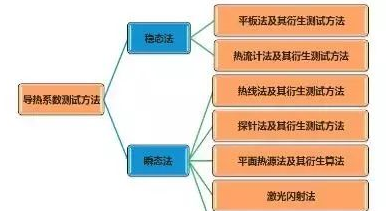
導熱系數測試方法-測試標準
針對材料導熱系數測試,除了需要有對應的測試方法和測試設備,還需要有對用的標準來規范測試方法、測試過程、測試條件、測試樣品、測試范圍等信息。
在材料導熱系數的測試領域,常用的導熱系數測試標準主要采用美國材料試驗協會(ASTM)的 ASTM-D5470,ASTM-E1461,ASTM-E1530,ASTM C518-04等。
熱膨脹分析
熱膨脹系數
物體的體積或長度隨溫度的變化而膨脹的現象稱為熱膨脹。其變化能力以等壓(p一定)下,單位溫度變化所導致的長度、面積或體積的變化,即熱膨脹系數表示。熱膨脹的本質是晶體點陣結構間的平均距離隨溫度變化而變化。材料的熱膨脹通常用線膨脹系數或者體膨脹系數來表述。
熱膨脹系數檢測意義熱膨脹系數是材料的主要物理性質之一,它是衡量材料的熱穩定性好壞的一個重要指標。
熱膨脹系數的影響因素
化學礦物組成:熱膨脹系數與材料的化學組成、結晶狀態、晶體結構、鍵的強度有關。組成相同,結構不同的物質,膨脹系數不相同。通常情況下,結構緊密的晶體,膨脹系數較大;而類似于無定形的玻璃,往往有較小的膨脹系數。鍵強度高的材料一般會有低的膨脹系數。
相變:材料發生相變時,其熱膨脹系數也要變化。純金屬同素異構轉變時,點陣結構重排伴隨著金屬比容突變,導致線膨脹系數發生不連續變化。
合金元素對合金熱膨脹有影響簡單金屬與非鐵磁性金屬組成的單相均勻固溶體合金的膨脹系數介于內組元膨脹系數之間。而多相合金膨脹系數取決于組成相之間的性質和數量,可以近似按照各相所占的體積百分比,利用混合定則粗略計算得到。
織構的影響:單晶或多晶存在織構,導致晶體在各晶向上原子排列密度有差異,導致熱膨脹各向異性,平行晶體主軸方向熱膨脹系數大, 垂直方向熱膨脹系數小。
內部裂紋及缺陷也會對熱膨脹產生影響
熱膨脹系數的檢測標準:
GB/T 34183-2017 建筑設備及工業裝置用絕熱制品 熱膨脹系數的測定
GB/T 3074.4-2016 石墨電極熱膨脹系數(CTE)測定方法
GB/T 16920-2015 玻璃 平均線熱膨脹系數的測定
GB/T 28194-2011 玻璃 雙線法線熱膨脹系數的測定
GB/T 25144-2010 搪玻璃釉平均線熱膨脹系數的測定方法
GB/T 16535-2008 精細陶瓷線熱膨脹系數試驗方法 頂桿法
密度測試
真密度(True Density)是相對于顆粒群的表觀密度和堆密度而言的,指材料在絕對密實的狀態下單位體積的固體物質的實際質量,即去除內部孔隙或者顆粒間的空隙后的密度。真密度是粉體材料最基本物理參數,也是測定微粉顆粒分布等其他物理性質必須用到的參數。真密度數值大小決定于材料化學組成及純度,其值直接影響材料質量、性能及用途,對其測定有重要意義。
真密度測試的方法:常用的測定真密度的方法主要是氣體容積法(加壓法)和浸液法(比重瓶法)兩種。所謂氣體容積法是根據氣體在密閉容器中遵守質量守恒定律,由測得的壓力來確定待測樣品(粉體)的體積,再由樣品的質量來最終測量樣品的密度。所謂浸液法,是根據阿基米德原理,測定粉體的真體積,再由粉體的質量計算其真密度。
浸漬法(比重瓶法):適用:適用于粉料,片料,粒料或制品部件的小切此。原理:該法測定粉體真密度基于阿基米德原理。方法:將待測粉末浸人對其潤濕而不溶解的浸液中,抽真空除氣泡,求出粉末試樣從已知容量的容器中排出已知密度的液體,就可計算所測粉末的真密度。
氣體容積法(氦氣加壓置換法):適用:適用于各類粉體、片狀、塊狀材料,尤其適合于多孔材料。原理:以氣體取代液體測定樣品所排出的體積。氣體能滲入樣品中極小的孔隙和表面的不規則空陷,因此測出的樣品體積更接近樣品的真實體積,從而可以用來計算樣品的密度,測試值更接近樣品的真實密度。方法:將試料置于真密度測試儀中,用氦氣作介質,在測量室逐漸加壓到一個規定值,然后氦氣膨脹進入膨脹室內,兩個過程的平衡壓力由儀器自動記錄,根據質量守恒定律,通過標準球校準測量室和膨脹室的體積后,再確定試料的體積,計算出真密度。
其他密度測試方法:排水法測密度--阿基米德原理、振實密度(堆密度)、松裝密度。
粒度測試--Zeta、激光粒度
粒度分析的基本概念
顆粒:具有一定尺寸和形狀的微小物體,是組成粉體的基本單元。它宏觀很小,但微觀卻包含大量的分子和原子。
粒度:顆粒的大小。
粒度分布:用一定的方法反映出一系列不同粒徑顆粒分別占粉體總量的百分比。
粒度分布的表示方法:表格法(區間分布和累積分布)、圖形法、函數法,常見的有R-R分布,正態分布等。
粒徑:顆粒的直徑,一般以微米為單位。
等效粒徑:指當一個顆粒的某一物理特性與同質球形顆粒相同或相近時,我們就用該球形顆粒的直徑來代表這個實際顆粒的直徑。
D10,累計分布百分數達到10% 所對應的粒徑值; D50,累計分布百分數達到50%時所對應的粒徑值;又稱中位徑或中值粒徑; D90,累計分布百分數達到90%時所對應的粒徑值。
測試方法:激光粒度儀和Zeta電位分析儀。
激光粒度儀:根據顆粒的衍射或者散射光的空間分布分析顆粒粒度的儀器,采用Furanhofer衍射及Mie散射理論,測試過程不受溫度變化、環境介質年度表面狀態等因素的影響。
Zeta電位儀:采用光子相關光譜法、電泳光散射及最新FST技術來分析的一款納米粒度儀和zeta電位一體機,并可測定固體以及高濃度懸浮液的zeta電位。該儀器采用了高靈敏度測量技術,同時滿足低濃度和高濃度樣品納米粒度與zeta電位分析的要求,可檢測的粒徑范圍為0.6nm-10?m、濃度范圍為0.00001%-40%的粒徑。
激光粒度和Zeta粒度測試的區別:
激光粒度適用于微米級粒徑的樣品;Zeta粒度(納米粒度)適用于納米級粒徑樣品。激光粒度所需樣品量多,需要將樣品分散到150ml溶劑中,而且測試需要達到一定遮光度,就要求樣品量多一些;而Zeta只需要分散在3ml的溶劑中,很低的濃度就可以。
其他粒度測量方法
篩分法按照被測試樣的粒徑大小及分布范圍,將大小不同篩孔的篩子疊放在一起進行篩分,收集各個篩子的篩余量,稱量求得被測試樣以重量計的顆粒粒徑分布。
沉降法(重力沉降法、離心沉降法)根據不同粒徑的顆粒在液體中的沉降速度不同測量粒度分布的一種方法。它的基本過程是把樣品放到某種液體中制成一定濃度的懸浮液,懸浮液中的顆粒在重力或離心力作用下將發生沉降。大顆粒的沉降速度較快,小顆粒的沉降速度較慢。斯托克斯Stokes 定律是沉降法粒度測試的基本理論依據。
電阻法(庫爾特顆粒計數器)根據小孔電阻原理,又稱庫爾特原理,測量顆粒大小的。
電鏡法:SEM、TEM;1nm~5μm范圍。適合納米材料的粒度大小和形貌分析。
超聲波法超聲波發生端(RF Generator)發出一定頻率和強度的超聲波,經過測試區域,到達信號接收端(RF Detector)。當顆粒通過測試區域時,由于不同大小的顆粒對聲波的吸收程度不同,在接收端上得到的聲波的衰減程度也就不一樣,根據顆粒大小同超聲波強度衰減之間的關系,得到顆粒的粒度分布。
透氣法粉末樣品的氣體透過能力與粉末的比表面有關,籍此求出樣品的比表面積并由此得到顆粒的平均粒度。
孔徑、開孔率及比表面積
BET測試
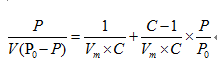
BET比表面積就是:Brunauer、Emmett和Teller三位科學家的首字母組合為BET。BET吸附指的是多層吸附,這也是材料的實際吸附過程。BET比表面積測試可用于測顆粒的比表面積、孔容、孔徑分布以及氮氣吸附脫附曲線。
P: 氮氣分壓P0: 吸附溫度下,氮氣的飽和蒸汽壓V: 樣品表面氮氣的實際吸附量Vm: 氮氣單層飽和吸附量C : 與樣品吸附能力相關的常數
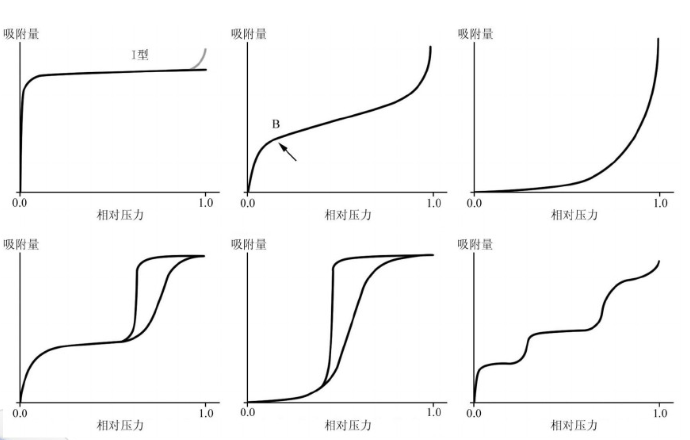
IUPAC提出的吸附等溫線分類
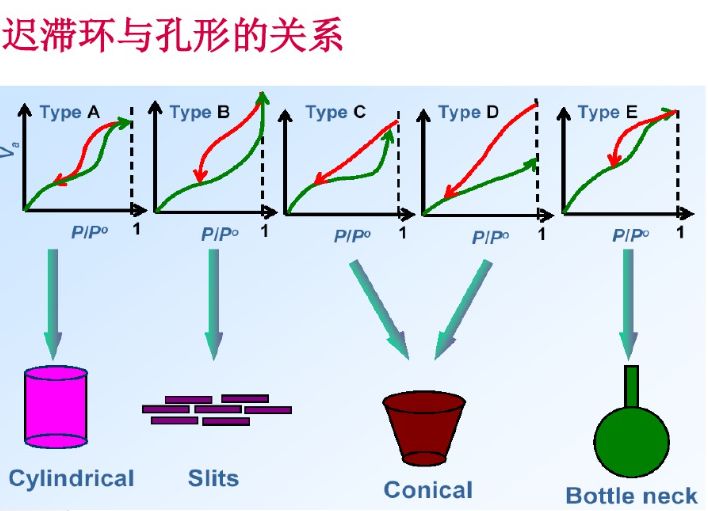
BET公式的使用范圍
通常情況下,BET公式只適用于處理相對壓力(p/p0)約為0.05~0.35之間的吸附數據。這是因為BET理論的多層物理吸附模型限制所致。當相對壓力小于0.05時,不能形成多層物理吸附,甚至連單分子物理吸附層也遠未建立,表面的不均勻性就顯得突出;而當相對壓力大于0.35時,毛細凝聚現象的出現又破壞了多層物理吸附。
壓汞測試
壓汞法(Mercury intrusion method)是指用來測定樣品中的過渡氣孔和宏觀氣孔的孔徑和它們的分布。壓汞法是依靠外加的壓力使汞克服表面張力進入樣品氣孔來測定樣品的氣孔孔徑和氣孔分布。外加壓力增大,可使汞進入更小的氣孔,進入樣品氣孔的汞量也就愈多。當假設樣品氣孔為柱形時,根據汞在氣孔中的表面張力與外加壓力平衡的原理,可以得到樣品孔徑的計算方法。
原理:假設多孔材料是由大小不同的圓桶形毛細管所組成,根據毛管內液體生姜原理,水銀所受壓力P和毛管半徑r的關系如下:(根據施加壓力,即可得到對應的孔徑尺寸)
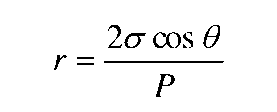
應用:主要針對中孔和大孔材料,測試顆粒的分布、顆粒間及顆粒內部的孔隙率、孔彎曲率、滲透性、孔喉比、分形特性和可壓縮性能等;結合BET法可對樣品孔結構進行整體分析。
壓電測試--壓電常數、彈性常數、介電常數、機械耦合系數
壓電檢測法是一種利用石英晶體微天平(QCM)進行測量的電化學檢測法。QCM是通過測定振子的基頻共振頻率的變化來檢測微小的質量變化,檢測靈敏度可以達到“納克”級且受實驗條件的干擾較小,在檢測DNA的固定、雜交等方面有廣泛的應用前景。
以寡核苷酸包覆的金納米粒子為放大探針,通過靶標分子與固定在QCM電極上的探針形成探針-靶-金納米粒子探針的三明治結構來進行信號檢測。與核酸堿基相比,金納米粒子的質量較大,因此得到的振蕩頻率的變化也較大,從而提高了QCM檢測的靈敏度。采用這種方法可簡便、快捷地檢測濃度在nmol/L級以下的靶標核苷酸。
霍爾效應、UPS--半導體參數
霍爾效應測試
在一個磁場中放置一塊導體,在垂直于磁場的方向上施加一個電流時,載流的帶電粒子會在磁場力作用下發生偏轉,導體橫向兩側的電荷會重新分布,從而產生一個內部的電場。這個現象便是霍爾效應,由此產生的導體兩端的電勢差通常稱為霍爾電壓(Hall voltage)。這一現象于1879年由美國科學家 Edwin Hall 發現,由此得名。
自旋霍爾效應的神奇之處在于,它甚至不需要外界磁場的幫助。這給了科學家一個不需通過磁場、只通過電流就能控制電子自旋的新思路。電子形成電流的過程中,很容易經歷隨機碰撞從而耗散能量。利用電子自旋和電流的規律,我們也許能夠指揮電子進行有序的運動,從而減低能耗。自旋霍爾效應有可能會在不久的將來應用到計算機的芯片制造中,設計出下一代的性能更優越的芯片。
應用:霍爾效應測試儀,是用于測量半導體材料的載流子濃度、遷移率、電阻率、霍爾系數等重要參數,而這些參數是了解半導體材料電學特性必須預先掌控的,因此是理解和研究半導體器件和半導體材料電學特性必備的工具。
UPS測試
紫外光電子能譜(UltravioletPhotoelectron Spectroscopy,簡稱UPS)兩個分支體系。Tunner 等人所發展的紫外光電子能譜,它的激發源在屬于真空紫外能量范圍,可以在高能量分辨率(10~20meV)水平上探測價層電子能級的亞結構和分子振動能級的精細結構,是研究材料價電子結構的有效方法。
紫外光電子能譜的測量原理:UPS測量的基本原理與XPS相同,都是基于愛因斯坦光電定律。
紫外光電子能譜的分析方法
紫外光電子能譜通過測量價層電子的能量分布從中獲得有關價電子結構的各種信息,包括材料的價帶譜、逸出功、VB/HOMO位置以及態密度分布等。
我們知道,在分析XPS譜圖時確定譜峰的位置至關重要。待測樣品中所含元素以何種化學態的形式存在,最主要的判據是化學位移,然而對以能帶結構為主要研究對象的UPS譜圖來說,除了需要對譜結構本身進行仔細辨識外,還要對譜的端邊進行精確標定。通常,半導體材料的 EF位于帶隙之間,它與價電子所能填充的最高能量位置-價帶頂(VBM)之間有一個未知的能量差。對于p型半導體材料,該能量差可以非常小,而對于n型半導體材料則可以大到與禁帶寬度Eg相當。而且由于半導體材料受表面態影響會在近表面處發生能帶彎曲,因此EF相對于 VBM的位置會隨表面處理條件的改變而變化,這在解析譜圖時需要考慮。
紫外光電子能譜的應用
最初,高分辨的UPS能譜儀主要用來測量氣態分子的電離電位,研究分子軌道的鍵合性質以及定性鑒定化合物種類。后來UPS越來越多地應用于廣延固體表面研究。固體的物理和化學性質與它們的能帶結構密切相關,廣延固體中的價電子結構較分子材料中單個原子或分子的價電子結構復雜得多。
目前,紫外光電子能譜是研究固體能帶結構最主要的技術手段之一。采用UPS研究固體表面時,由于固體的價電子能級被離域的或成鍵分子軌道的電子所占有,從價層能級發射的光電子譜線相互緊靠,因價電子能級的亞結構、分子振動能級的精細結構等疊加成帶狀結構,因此得到的光電子能量分布并不直接代表價帶電子的態密度,而應包括未占有態結構的貢獻,即受電子躍遷的終態效應影響,如自旋-軌道耦合,離子的離解作用,Jahn-Teller效應,交換分裂和多重分裂等。
未來科學技術的發展,對各種無機非金屬材料,尤其是對特種新型材料提出更多更高的要求。材料學科有廣闊的發展前景,復合材料、定向結晶材料、增韌陶瓷以及各種類型的表面處理和涂層的使用,將使材料的效能得到更大發揮。
免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。
相關文章

官方微信
《腐蝕與防護網電子期刊》征訂啟事
- 投稿聯系:編輯部
- 電話:010-62316606-806
- 郵箱:fsfhzy666@163.com
- 腐蝕與防護網官方QQ群:140808414
點擊排行
PPT新聞

“海洋金屬”——鈦合金在艦船的
點擊數:8208

腐蝕與“海上絲綢之路”
點擊數:6533