
計算干貨 | 基于理論與計算化學Gaussian軟件研究緩蝕劑的緩蝕機理
2017-08-30 12:23:48
作者:本網整理 來源:材料人
分享至:


(1)基于Gaussian軟件研究緩蝕劑
為了研究不同緩蝕劑的緩蝕機理,我采用了Gaussian軟件,在6-31G++(d,p)基組水平上,對所研究的緩蝕劑苯并三唑和硫脲分子進行了幾何構型優化,并進行頻率分析,確保所得結構均為勢能面上的極小點無虛頻。
通過使用ChemOffice軟件中的Chemdraw軟件,繪制了兩種緩蝕劑苯并三唑和硫脲分子的結構式,如圖27所示。
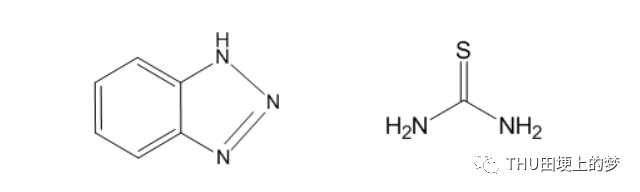
圖27. 兩種緩蝕劑苯并三唑(左)和硫脲(右)分子的結構式
在同一基組水平上,分別計算了苯并三唑和硫脲緩蝕劑分子的前線軌道分布和Fukui指數,用于分析分子的反應活性區域和吸附位點。
獲得相關參數:最高占據軌道HOMO和最低空軌道LUMO的能量及兩者之間的能量差,全局參量,如電離能、電子親和能、電負性、硬度、偶極距、總能量、分子體積和電子分布等參量。
緩蝕劑分子的電負性和硬度的關系式如下:
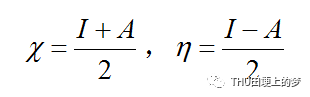
從緩蝕劑分子中轉移到金屬的電子數是根據從所獲得的和的值計算得到的,計算公式如下:
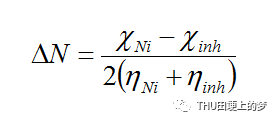
其中使用理論值,由于它比中性原子更加柔軟,因此,通過假設金屬的硬度為一個整體,即I=A。
(2)緩蝕劑分子的優化幾何構型
密度泛函理論DFT是一種經濟、高效的量子化學計算方法,它可以提供足夠精確的分子幾何構型和電子分布等信息。
首先我對這兩種緩蝕劑苯并三唑和硫脲分子進行分子結構優化。圖28是硫脲分子的最優構型。所涉及的量子化學參數均是在分子最優構型的基礎上計算所得。
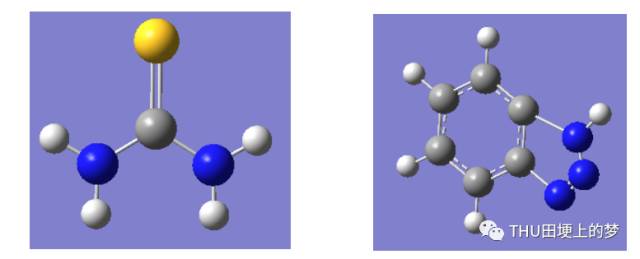
圖28. 硫脲分子(左)和苯并三唑分子(右)的最優構型
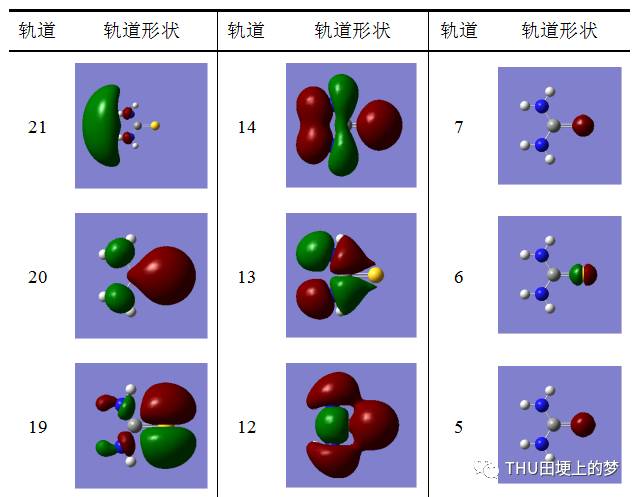
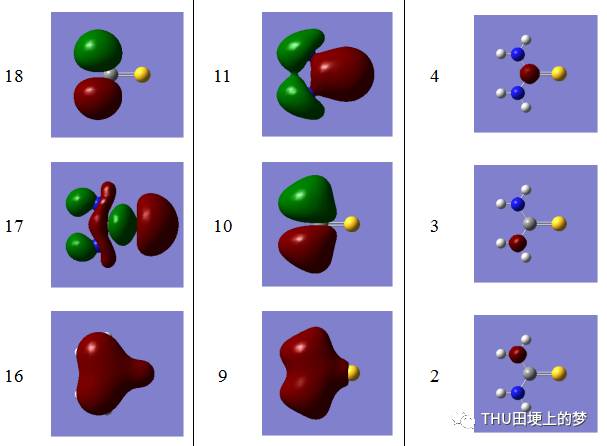
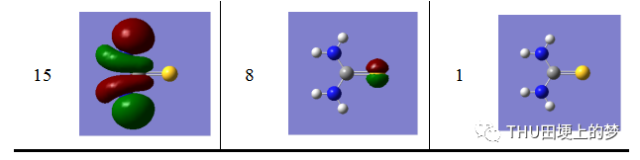
表13是使用Gaussian軟件計算得到的硫脲分子的分子軌道數目和形狀。
表13. 硫脲分子的分子軌道形狀
(3)緩蝕劑全局反應活性
我對所研究的緩蝕劑分子做了模擬計算,并得出相應的量子化學參數,可以用于分析緩蝕劑分子的反應活性和選擇性能。
量子化學的前線軌道理論認為反應物間的相互作用僅發生在分子的前線軌道之間,分子中的EHOMO是分子給電子能力的大小,分子的EHOMO越高,則該分子越易提供電子參與親核反應。分子的EHOMO是與分子的電子親和能有關,其值越低,該分子則有較強的接受電子的能力。因此分子的最高占據軌道HOMO和最低空軌道LUMO是分析緩蝕劑在金屬表面吸附行為的重要依據。
圖為無溶劑狀態下兩種緩蝕劑苯并三唑和硫脲分子的HOMO和LUMO分布圖。
表14是兩種緩蝕劑苯并三唑和硫脲分子量化計算的結構參數。
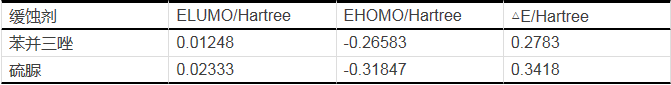
從表14中可以看出,從EHOMO的大小來看,苯并三唑的EHOMO為-0.26583 Hartree,硫脲的EHOMO為-0.31847 Hartree,EHOMO越大越好,說明苯并三唑的緩蝕效率最高,與極化曲線得出的結論相一致。
從ELUMO來看,苯并三唑的ELUMO為0.01248 Hartree,硫脲的ELUMO為0.02333 Hartree,ELUMO越小越好,苯并三唑的緩蝕效率最好。再從△E來看,苯并三唑的△E為0.2783 Hartree,硫脲的△E為0.3418 Hartree,△E越小越好,苯并三唑的緩蝕效率最好。從這三組參數的結果中我們可以確定出苯并三唑緩蝕劑效果比硫脲緩蝕劑效果好。
(4)結論
1. 苯并三唑的緩蝕效率優于硫脲的緩蝕效率,其中,苯并三唑屬于以抑制陽極為主的混合型緩蝕劑,硫脲屬于以抑制陰極為主的混合型緩蝕劑。
2. 兩種緩蝕劑苯并三唑和硫脲分子在鋁和鎳表面的吸附均服從Langmuir等溫式,吸附過程是放熱過程,屬于物理吸附。因此理論上,在所研究的溫度范圍內兩種緩蝕劑苯并三唑和硫脲分子的緩蝕性能隨溫度的升高而降低。
3. 運用量子化學理論和分子設計等先進科學技術在理論上預測和計算緩蝕劑,在特定環境中的緩蝕效率和緩蝕機理等。在某種程度上,可以減少實驗時測試的工作量,有利于合理選擇緩蝕劑,克服實驗的盲目性。
更多關于材料方面、材料腐蝕控制、材料科普等方面的國內外最新動態,我們網站會不斷更新。希望大家一直關注中國腐蝕與防護網http://www.ecorr.org
責任編輯:王元
投稿聯系:編輯部
電話:010-62313558-806
中國腐蝕與防護網官方 QQ群:140808414
免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。
相關文章

官方微信
《中國腐蝕與防護網電子期刊》征訂啟事
- 投稿聯系:編輯部
- 電話:010-62316606-806
- 郵箱:fsfhzy666@163.com
- 中國腐蝕與防護網官方QQ群:140808414
點擊排行
PPT新聞

“海洋金屬”——鈦合金在艦船的
點擊數:8125

腐蝕與“海上絲綢之路”
點擊數:6461