
電化學腐蝕
2021-07-28 14:04:38
作者: 工業小南點 來源: 工業小南點
分享至:


第二章 腐蝕的基本原理
電化學腐蝕
電化學腐蝕是金屬材料與電解質溶液互相接觸時,在界面上發生有自由電子參加的廣義氧化和還原反應,使接觸面的金屬變為離子而溶解或生成穩定化合物的過程,是以金屬為陽極的腐蝕電池過程。金屬在電解質溶液(包括大氣腐蝕情況下的薄水膜)和熔鹽中的腐蝕過程是電化學腐蝕過程,電解質溶液和熔鹽的共同特征是它們都是離子導體,依靠帶電荷離子的活動而導電。
2.3.1 電化學腐蝕過程的基本原理
本質上,金屬電化學腐蝕過程與金屬的化學腐蝕過程一樣,都是氧化還原反應,即金屬原子被氧化(失去價電子),而某一氧化劑被還原。但這兩類腐蝕過程的氧化還原的進行方式又有重大區別。
在化學腐蝕過程的情況下,氧化還原過程只有在反應粒子(氧化劑的分子或原子和金屬的原子)相互直接碰撞的過程中才能發生。所以,在氧化還原反應中的氧化過程和還原過程兩者不僅必須在同時,而且必須在同一個碰撞點發生。
電化學腐蝕過程則不然,雖然氧化過程和還原過程是必須同時進行的,但氧化劑的粒子不必直接同被氧化的那個金屬原子碰撞,而可以在金屬表面上的其他部分得到電子。
這就是說,在電化學腐蝕過程中,整個腐蝕反應分成兩個既是互相聯系又是相對獨立的半反應分別同時進行的。如鋅在除氧的硫酸中腐蝕時兩個半反應:
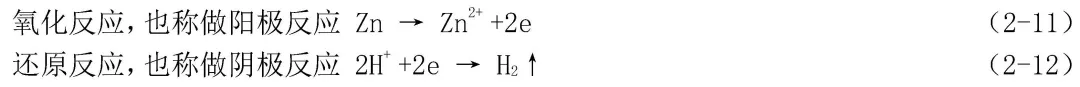
在這兩個反應方程式所表示的腐蝕反應中,氧化劑是H+,作為去極化劑,在腐蝕反應中被還原成氫分子。
由于電化學腐蝕過程的這一基本特點-兩個半反應在空間上的可分性,使得兩個半反應可以各自在最有利于它們進行的地點進行,從而也使得整個腐蝕反應可以在阻力最小的條件下進行。
2.3.2 發生電化學腐蝕的熱力學條件
一個腐蝕反應是否會發生,同任何其他過程一樣,取決于這個反應進行時整個體系的自由能是否降低。如從反應體系的始態轉變為反應體系的終態,自由能降低,則這個反應就能夠自發地進行。對于一個電化學腐蝕反應來說,自由能變化ΔG可以用式(2-13)表示:
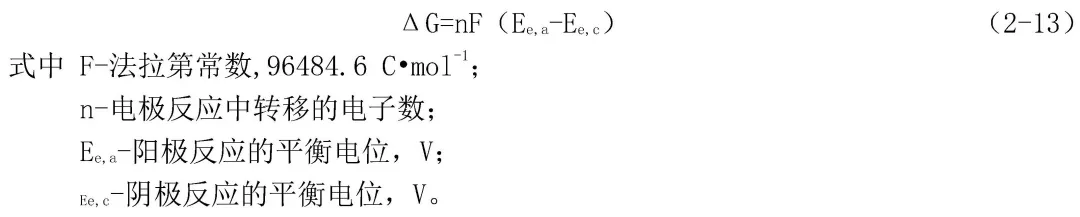
由式(2-11)看出,只有當時Ee,a-Ee,c>0 腐蝕反應才能發生。
如果一個電極反應(半反應)處于平衡,相應的電位叫做這個電極反應的平衡電位Ee,Ee與參與這個電極反應的物質活度之間的關系可以用能斯特(Nernst)方程式表達:
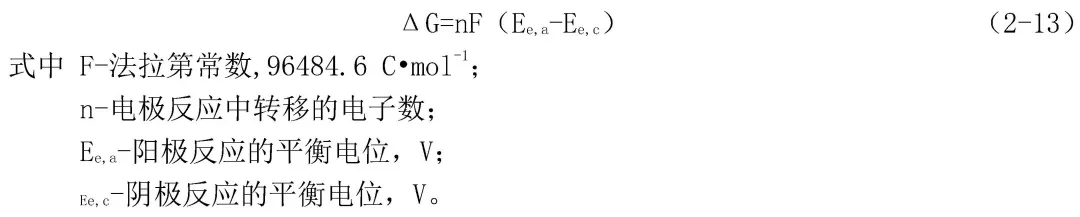
標準電極電位是當參加電極反應的物質處于標準狀態下,即溶液中該種物質的離子活度為1、溫度為298.15 K、氣體分壓為101325 Pa (1atm) 時,電極的平衡電極電位,用Eo表示。國際上規定標準氫電極電位為零,在沒有特殊說明條件下,其他電極的電極電位都是以標準氫電極為基準。通常用金屬的標準電極電位可以近似地判斷它們的熱力學穩定性。
E0可以在相關電化學手冊中查找,根據能斯特方程式計算電化學腐蝕過程的平衡電位Ee,a和Ee,c,按式(2-11)判斷這個電化學過程是否可能發生。
2.3.3 溶液的pH值對電化學腐蝕的影響
在水溶液中,常溫下[H+][OH-]=10-14,這與電化學腐蝕過程有著密切的關系,在水溶液中的電化學腐蝕過程的陰極反應一般都是H+或O2的去極化過程,兩個電極反應都涉及H+或OH-,反應式為:

溶液的pH值還會影響到陽極反應的形式和反應產物,如在pH值較高的溶液中,還可以進行如下陽極反應:
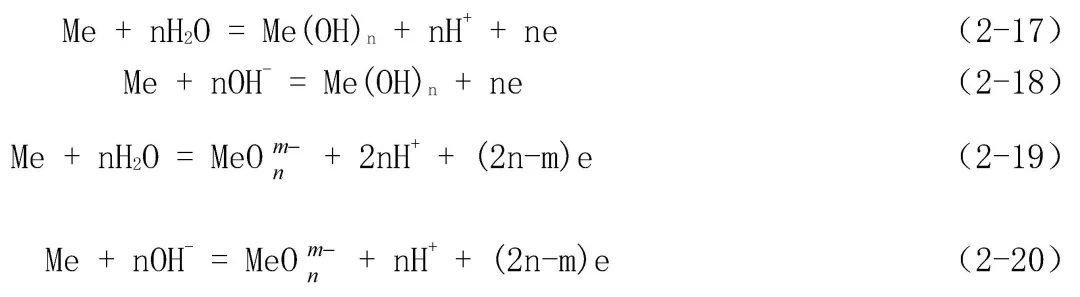
2.3.4 E-pH圖(pourbAlx diagram)
在金屬腐蝕過程中,電位是控制金屬離子化過程的因素,pH值是控制膜穩定性的因素。應用這2個因素,以電位(相對于標準氫電極)為縱坐標、以pH值為橫坐標的電化學相圖,又稱布拜圖。據此,可從熱力學上判斷某種金屬在給定的電位和pH值條件下是否會發生腐蝕及其平衡狀態。它可以指出金屬腐蝕的傾向,但并不能指示腐蝕速度。目前已將幾乎全部元素的這種平衡圖研究完成,近年來,這些平衡圖已越來越多地在金屬腐蝕、電化學以及其它有關領域里得到應用。
根據電化學反應的原理,可以將與金屬在水溶液中的腐蝕過程有關的反應概括為3種類型:
① 化學價有變化(得到或失去電子,即還原與氧化),但沒有H+或OH-參與的反應,這一類反應的平衡只與金屬的電位E有關,而與溶液的pH值無關。如:
Me = Men+ + ne (2-21)
② 化學價沒有變化,但有H+或OH-參與的反應,這一類反應的平衡只與溶液的pH值有關,而與金屬的電位E無關。如:

③ 化學價有變化,而且有H+或OH-參與的反應,這一類反應的平衡既與金屬的電位E有關,又與溶液的pH值有關。如:
Me + nOH- = Me(OH)n↓ + ne(2-24)
以電位為縱軸,溶液的pH值為橫軸,將一種金屬在水溶液中與腐蝕過程有關的反應的平衡值繪制平衡線,則在其他參與條件保持不變的情況下,可以得到3種類型的平衡線:
① 平行于橫軸的水平線,平衡只與溶液pH值有關;
② 平行于縱軸的垂直線,平衡與只金屬的電位E有關;
③ 傾斜線,平衡既與金屬的電位E有關,又與溶液的pH值有關。
Fe的E-pH圖,如圖2-6。
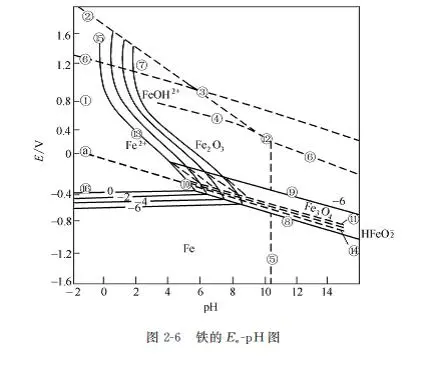
利用E-pH圖,可以根據金屬的電位和溶液的pH值來判斷:
① 腐蝕區:如果金屬的電位E和溶液的pH值的觀察點落在金屬離子穩定的區域,腐蝕過程就可能發生。
如果發生腐蝕反應,是以O2為去極化劑,還是H+為去極化劑,在E-pH圖中有兩條傾斜的平行的虛線。上面一條是O2還原成為OH-的反應(Po2=101kPa)的平衡線,下面一條是H+還原為H2的反應(PH2=101kPa)的平衡線。如金屬的電位和溶液的pH值的觀測點落在下面那條虛線的下面,腐蝕反應就既能以O2為去極化劑,也能以H+為去極化劑。如果觀測點位于這兩條虛線之間,那么腐蝕過程只能以O2為去極化劑。
② 鈍化區:如果金屬的電位和溶液的pH值的觀測點落在金屬的氧化物或金屬的氫氧化物穩定的區域,就有可能生成腐蝕產物膜或鈍化膜。這是材料獲得陽極保護的依據,在此區域內,金屬是否遭受腐蝕,取決于所生成的固態膜是否具有保護性。
③ 穩定區(非腐蝕區):在該區域內,E和pH值的變化不會引起金屬的腐蝕,即在熱力學上,金屬處于穩定狀態。這些耐蝕性不同的區域的具體位置取決于選用的臨界條件,隨臨界條件不同而不同,常用溶液中金屬離子或金屬配合離子的活度為10-6作為臨界條件,這是材料獲得陰極保護的依據,一般說來,只要將金屬的電位降低到低于金屬離子活度為10-6的平衡線,金屬就能得到充分的陰極保護。
2.3.5 金屬電化學腐蝕過程的基本動力學規律
金屬電化學腐蝕的整個腐蝕反應過程,包括4大步驟:去極化劑到達金屬表面的傳質過程;陽極反應過程;與陽極反應過程同時進行的去極化劑還原的陰極反應過程;腐蝕產物離開金屬表面或轉化為其他化合物的過程,每一大步驟中都包括一系列中間步驟。在這4大步驟中,前3個步驟無論哪一個步驟進行困難都會影響整個腐蝕反應的速度,稱為控制步驟,整個腐蝕反應的速度主要由控制步驟的進行速度控制。在研究腐蝕過程時,很重要的是要弄清這個腐蝕過程的速度控制步驟,知道了一個腐蝕過程的速度控制步驟,就可以設法對控制步驟施加影響,使它的進行更加困難,就易于使整個腐蝕過程的速度降低。
① 傳質過程。影響去極化劑到達金屬表面的傳質過程速度的主要因素有:溶液與金屬表面之間的相對運動速度、去極化劑在溶液中的含量、溶液的溫度和黏度。一般來說,溶液與金屬表面的相對運動速度越大、溶液中去極化劑的含量越高、溶液的溫度越高和溶液的黏度越小傳質過程就進行得越快。例如,鋼鐵在海水中的腐蝕過程是以溶解在海水中的O2 為去極化劑的,在靜止的海水中O2 向鋼鐵表面的擴散過程是控制步驟,但如果海水與鋼鐵表面之間以很高的速度相對運動,O2的擴散過程就大為加速,以至于這個傳質過程可以不再是整個腐蝕過程的控制步驟。所以靜止條件下的腐蝕試驗結果與溶液流動條件下的會有很大的不同。另外還要注意,影響傳質過程速度的各因素中有些是互相影響的,特別對溫度這個因素要加以注意,溫度升高會使擴散過程容易進行,而且溶液的黏度一般也是隨著溫度的升高而降低,所以,一般溫度升高,腐蝕速率會上升;但氣體在溶液中的溶解度是隨著溫度的升高而降低,如果去極化劑是氣體,而且進行腐蝕體系是敞開的,溫度升高使溶液中溶解氣體的含量降低,因此,腐蝕速率也相應下降。
② 陽極或陰極反應過程。這是一個涉及電子得失的過程。腐蝕過程中的陽極反應過程是金屬原子失去價電子而成為金屬離子或化合物的過程,金屬的腐蝕破壞是它的直接結果,其速度與金屬的電位有關。在金屬的電位E等于陽極反應的平衡電位Ee,a時,陽極反應處于平衡,宏觀的反應速度為零。只有當金屬的電位E偏離平衡點位形成過電位,陽極或陰極反應的宏觀速度才大于0。電極反應動力學規律為:
a.過電位是電極上有電流通過時的電極電位與其初始電位的差值,η=E-E平。過電位反映了極化作用的大小。過電位越大,極化作用越大。對于腐蝕原電池,極化增加原電池對外做功的能力大大下降,腐蝕速率下降。也就是說,極化有利于金屬的電化學防護,但不利于原電池放電。用初始電位計算得到的腐蝕速率要比實際腐蝕速率大1-2個數量級,就是因為極化作用的影響。
b.η>0,電極反應按陽極反應方向進行;η<0,電極反應按陰極反應方向進行。
c.過電位的數值越大,在金屬的表面狀態沒有改變的條件下(如,金屬表面上沒有生成鈍化膜或其他腐蝕產物膜)陽極反應的速度就越大。所以,在金屬表面狀態不變的情況下,凡是能使金屬的電位E提高或平衡電位Ee,a降低,從而使得陽極反應過電位增大的因素,都會加速金屬的陽極溶解反應,反之亦然。如Fe在酸中腐蝕時的電位比Zn在酸中腐蝕的電位高,如果在酸中這兩種金屬發生“電接觸”,那么Zn的電位將會升高而Fe的電位將會降低直至兩者接近相同,故Zn的陽極溶解速度將因這種電接觸而增大,Fe的陽極溶解速度則降低。又如,溶液中存在著能同金屬離子絡合的物質,就使金屬陽極溶解反應的平衡電位降低,從而加速金屬的陽極溶解速度。
d.由于電極反應發生于金屬表面,是一種表面反應,故其他物質在金屬表面上的吸附會影響陽極反應的速度。有的會加速金屬的陽極溶解速度,如Cl-;有的物質吸附在金屬表面上后,則使陽極反應減速,如某些緩蝕劑。許多有機胺在金屬表面上的吸附,能降低H+還原的反應速度。
e.陽極鈍化。陽極極化可以使金屬表面形成完整的能阻礙金屬離子穿過的表面膜,陽極溶解過程就會受到極大抑制。特別是一些金屬,例如黑色金屬,在一些介質中電位E提高到相當高的數值,金屬的表面狀態會發生變化,生成一種很薄,但金屬離子很難穿過的氧化膜。此時陽極溶解速度可以降得很低,這種膜就稱為鈍化膜,生成鈍化膜的過程叫做鈍化過程。
f.陰極反應也是一種表面反應,金屬表面的性質對于反應有很大影響。例如H+ 還原為H2的反應,在Pt、Pd上很容易進行,在Cu、Fe表面就難一些,而在Pb、Hg等金屬表面上則就相當困難。
③ 腐蝕產物離開金屬表面或轉化為其他化合物的過程是電化學腐蝕反應已經完成以后的過程,但有時這一步驟進行的情況也會反過來影響前面3個步驟,從而影響整個腐蝕過程的速度。例如,如果腐蝕反應產物最終形成固體物質而覆蓋在金屬表面,就會使得前面的3個步驟進行困難,從而抑制腐蝕速度。或腐蝕產物形成疏松的沉積物,使金屬表面局部腐蝕環境發生變化,引起垢下腐蝕。
2.3.6 腐蝕電池
腐蝕原電池的原理與一般原電池的原理一樣,它只不過是將外電路短路的電池。
電化學腐蝕的特點是氧化過程和還原過程在空間上的可分,陽極反應和陰極反應的表面區域構成了腐蝕電池。腐蝕電池實質上是一個短路原電池,電流不對外作功,電子自耗于腐蝕電池內陰極還原反應中。腐蝕電池的構成以及陽極區和陰極區的分布情況對腐蝕破壞的形式有很大影響,腐蝕電池的形成可以使腐蝕過程以最有利于它進行的方式進行,所以它的形成都是使腐蝕加速,腐蝕破壞總是主要集中在陽極區,如果腐蝕電池是由大的陰極區和小的陽極區構成的,就會出現危險性較大的局部腐蝕的形式。
一個腐蝕電池必須包括陽極、陰極、電解質溶液和電路4個不可分割的部分。構成電池的3個必要條件為:
① 存在電位差,要有陰極、陽極存在;
② 有電解質溶液存在,溶液中有氧化劑(根本原因);
③ 在腐蝕電池的陰、陽極之間要有連續傳遞電子的回路。
腐蝕電池和一般丹尼爾電池的區別在于:
① 不是一種可逆電池;
② 不能將化學能轉化為電能,氧化還原反應所釋放的化學能全部以熱能方式散發;
③ 只能導致金屬材料破壞。
腐蝕電池的工作過程:
① 陽極過程:Me → Men+ + ne (2-25)
② 陰極過程:D + ne → Dne (2-26)
③ 電流流動:
金屬中:電子從陽極到陰極;
溶液中:陽離子從陽極向陰極移動、陰離子從陰極向陽極移動;
金屬/電解質界面電遷移,電子由低電位金屬或區域傳荷到電位高的金屬或區域,再轉移給氧化劑。
腐蝕電池的特點:
①陰、陽極區宏觀可分或不可分,或交替發生;陰極、陽極反應相對獨立,但又必須耦合,形成腐蝕電池;
②金屬的腐蝕集中出現在陽極區,陰極區只起傳遞電子的作用;ia=ic,無凈電荷積累;
③ 上述3個工作過程相互獨立,又彼此聯系;
④ 只要介質中存在氧化劑 (去極化劑),能獲得電子使金屬氧化,腐蝕就可發生;體系由不穩定到穩定,腐蝕過程是自發反應,并以最大限度的不可逆方式進行;
⑤ 腐蝕的二次產物對腐蝕影響很大;
⑥ 腐蝕電池不對外作功,是只導致金屬腐蝕破壞的短路原電池。
腐蝕電池可分為宏觀腐蝕電池、微觀腐蝕電池和亞微觀(10-100?)腐蝕電池。
要想使整個金屬的物理和化學性質、金屬各部位所接觸介質的物理和化學性質完全相同,使金屬表面各點的電極電位完全相同是不可能的。由于種種因素使得金屬表面的物理和化學性能存在著差異,使金屬表面上各部位的電位不相等,這些情況統稱為電化學不均勻性,它是形成腐蝕電池的基本原因。金屬表面的腐蝕電池都是微電池,金屬表面由微陰極和微陽極組成的眾多微電池是用目視難以分辨出電極的極性,但確實存在著氧化和還原反應過程的原電池。形成腐蝕微電池的主要原因有:
① 金屬表面電化學不均勻性,使金屬材料表面存在微小的電位高低不等的區域;
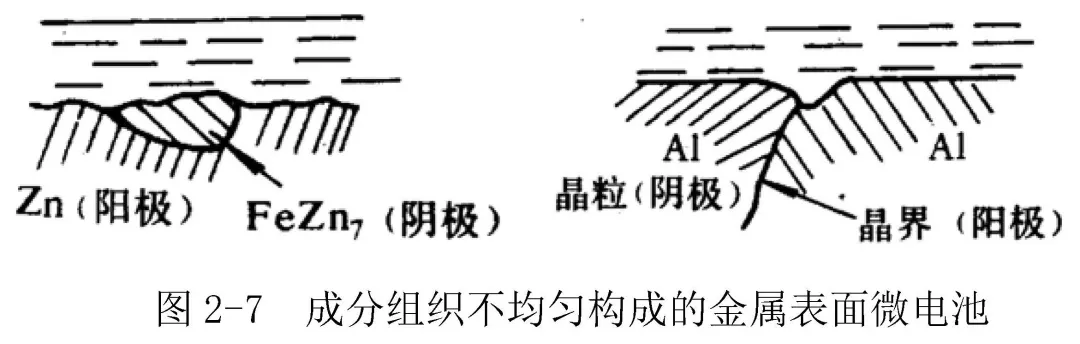
② 成分和組織不均勻引起的微電池;如:碳鋼中的滲碳體Fe3C,工業純鋅中的鐵雜質FeZn7,鑄鐵中的石墨等,晶粒-晶界腐蝕微電池,圖2-7。
③ 金屬表面物理狀態的不均勻性構成微觀電池。如應力分布不均勻或形變不均勻,導致腐蝕微電池,圖2-8。
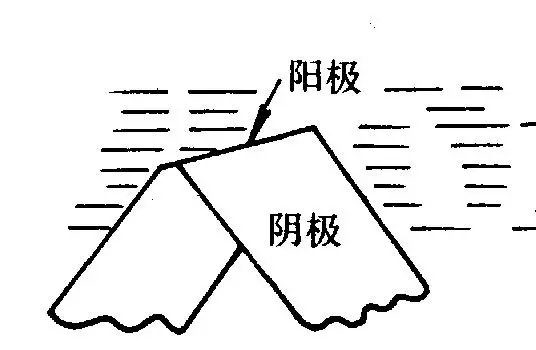
圖2-8 金屬表面物理狀態的不均勻性構成微觀電池
④ 金屬表面膜不完整構成微觀電池。金屬表面形成的鈍化膜或鍍覆的涂層存在孔隙或發生破損,裸露出金屬基體,金屬基體電位較負,鈍化膜或覆層的電位較正,金屬基體與鈍化膜或陰極性涂層構成微觀腐蝕電池,孔隙或破損處作為陽極而受到腐蝕,圖2-9。
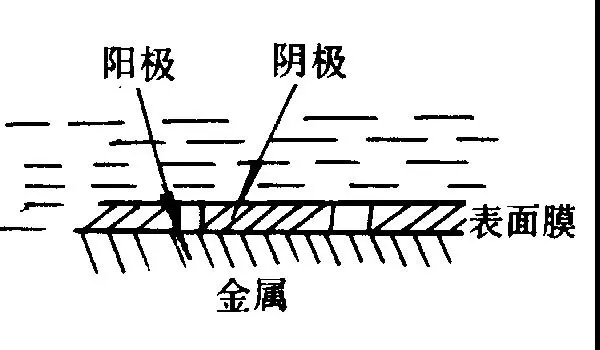
圖2-9 金屬表面膜不完整構成微觀電池
腐蝕體系的宏觀差異,還可以構成腐蝕宏電池,如異種金屬的接觸、介質的濃度差、介質或金屬的溫度差、沉積物分布、金屬構件的應力差等都可以構成腐蝕電池。
化學腐蝕與電化學腐蝕的區別見表2-1。

免責聲明:本網站所轉載的文字、圖片與視頻資料版權歸原創作者所有,如果涉及侵權,請第一時間聯系本網刪除。
相關文章

官方微信
《中國腐蝕與防護網電子期刊》征訂啟事
- 投稿聯系:編輯部
- 電話:010-62316606-806
- 郵箱:fsfhzy666@163.com
- 中國腐蝕與防護網官方QQ群:140808414
點擊排行
PPT新聞

“海洋金屬”——鈦合金在艦船的
點擊數:7130

腐蝕與“海上絲綢之路”
點擊數:5741